| Journal of Cancer Stem Cell Research (2015), 3:e1001 © 2015 Creative Commons. All rights reserved ISSN 2329-5872 DOI: 10.14343/JCSCR.2015.3e1001 http://cancerstemcellsresearch.com |
|
| Journal of Cancer Stem Cell Research (2015), 3:e1001 © 2015 Creative Commons. All rights reserved ISSN 2329-5872 DOI: 10.14343/JCSCR.2015.3e1001 http://cancerstemcellsresearch.com |
|
| Research Article | Open Access |
| Enhanced activation of STAT3 in ascites-derived recurrent ovarian tumors: inhibition of cisplatin-induced STAT3 activation reduced tumorigenicity of ovarian cancer by a loss of cancer stem cell-like characteristics | |
| Khalid Abubaker1,2,*, Ardian Latifi1,2,*, Emily Chan1,3, Rodney B. Luwor4, Christopher J. Burns5,6, Erik W. Thompson7, Jock K. Findlay1,3,8, and Nuzhat Ahmed1,2,3,8,* | |
| 1Women's Cancer Research Centre, Royal Women's Hospital, Victoria 3052, Australia, 2University of Melbourne, Department of Surgery, St Vincent's Hospital, Victoria 3065, Australia, 3Department of Obstetrics and Gynaecology, University of Melbourne, Victoria 3052, Australia, 4Department of Surgery, University of Melbourne, Royal Melbourne Hospital, Victoria 3052, Australia, 5Walter and Eliza Hall Institute of Medical Research, Victoria 3052, Australia, 6Department of Medical Biology, University of Melbourne, Vic 3010, Australia, 7Institute of Health and Biomedical Innovation and School of Biomedical Sciences, Queensland University of Technology, Australia, 8MIMR-PHI Institute of Medical Research, Victoria 3168, Australia. | |
| *contributed equally to the work | |
| *Corresponding author: Dr Nuzhat Ahmed, Women's Cancer Research Centre, Royal Women's Hospital, 20 Flemington Road, Parkville, Vic 3052, Australia. E-mail: nuzhata@unimelb.edu.au Received: December 30, 2014; Revised: January 29, 2015; Accepted: January 29, 2015 | |
Abstract: Chemotherapy resistance is a major obstacle for the treatment of ovarian cancer patients. Combination of drugs which can exert synergistic effect can be a promising strategy to overcome this resistance. In this study, we report significantly enhanced activation of Janus kinase 2 (JAK2) and its downstream target signal transducer and activation of transcription 3 (STAT3) in tumor cells isolated from the ascites of recurrent ovarian cancer patients (CR) compared to tumor cells isolated from the ascites of chemonaïve patients (CN). Enhanced activation of JAK2 and STAT3 in the tumor cells of recurrent patients coincided with the in vitro activation of JAK2 and STAT3 pathway in cisplatin-surviving ovarian cancer cell lines. This coincided with the emergence of cancer stem cell (CSC)-like characteristics in response to cisplatin treatment in ovarian cancer cell lines. Both cisplatin-induced JAK2/STAT3 activation and CSC-like characteristics were inhibited by a low dose JAK2-specific small molecule inhibitor CYT387 in vitro. Subsequent, in vivo transplantation of cisplatin and CYT387 in vitro treated ovarian cancer cells in mice resulted in a significantly reduced tumor burden compared to that observed in mice injected with cisplatin only-treated cells. In vitro analysis of tumor xenografts at the protein level demonstrated a loss of CSC-like (CD117 and Oct4) and tumorigenic (CA125) markers in cisplatin and CYT387-treated cell-derived xenografts, compared to cisplatin only-treated cell-derived xenografts. These results were consistent with a significantly reduced activation of JAK2 and STAT3 in cisplatin and CYT387-treated cell-derived xenografts compared to cisplatin only-treated cell derived xenografts. These data suggest that the inhibition of the JAK2/STAT3 pathway by the addition of CYT387 in combination with cisplatin may have important implications for ovarian cancer patients who are treated with platinum-based first line therapies.
Keywords: ovarian carcinoma, cancer stem cell, metastasis, ascites, chemoresistance, recurrence, JAK2/STAT3 pathway.
Ovarian cancer continues to be a major worldwide gynaecological malignancy with 15,000 deaths and 25,000 new cases diagnosed each year in US alone [1]. The cancer is extremely heterogeneous and manifests in multiple morphological forms within the major commonly recognised sub-types [2]. The treatment options for the majority of the four sub-types, that is serous, endometrioids, mucinuous and clear cell carcinomas are similar and have remained unchanged for the last two decades [3]. Thus, most ovarian cancer patients are treated with platinum or taxane-based chemotherapy, or a combination of both, which results in killing of tumor cells by affecting the DNA or cytoskeletal elements of the cells [4]. Although, these interventions initially provide a short disease-free progression period they generally result in the evolution of an aggressive and drug-resistant disease that ultimately results in disease progression and ultimately patients' death [5]. Hence, the discovery and development of targeted therapies that directly affect chemotherapy resistance and regrowth of drug-refractory tumors is urgently needed for the treatment of ovarian cancer patients.
With the recent advancement in gene expression profiling and the implementation of next generation DNA sequencing and RNA sequencing technologies, a broad heterogeneity in samples from patients diagnosed with the serous ovarian tumors has been identified [6, 7]. Although these tools have segregated tumors into broad subclasses, the reasons behind this enormous genetic variability are still unknown and as such no suitable targeted therapeutic options are available.
One important concept in cancer biology has gained considerable momentum in the past decade is the concept that supports the cancer stem cell (CSC) paradigm, a theoretical model that not only supports the existence of a small sub-population of tumor cells that drives the tumor growth and progression but also sustains cytotoxic therapeutic pressure and persists to support the re-growth of tumors [8]. Accordingly, it is reasonable to suggest that cancer will be best diagnosed and treated if knowledge of the events related to CSCs are unravelled. Hence, the development of personalised medicine will depend on the efficient implementation of DNA and RNA sequencing of the identified tumor specific CSCs in each of the respective tumor subtypes as well as the pathways regulating the survival of these CSCs.
We and others have recently demonstrated an association between chemoresistance and CSC-like phenotypes in ovarian cancer [9–12], and found chemoresistant recurrent ovarian tumors to be enriched in CSCs and stem cell pathway mediators, suggesting that CSCs may contribute to recurrent disease [13, 14]. CSCs have also been isolated from ovarian cancer cell lines based on their abilities to differentially efflux the DNA binding dye Hoechst 33342 [15]. This population of cells termed the ‘side population’ (SP) displayed the classical stem cell property in tumorigenicity assays. Other recent reports have shown the presence of CSCs in ovarian tumors in patients' ascites [16–20]. CSCs in these studies were reported to be resistant to conventional chemotherapy and were able to recapitulate in vivo the original tumor suggesting that these CSCs control self-renewal as well as metastasis and chemoresistance.
The JAK/STATs are well-characterised signalling kinases which are activated in response to growth factors and cytokines through the phosphorylation of tyrosine residues [21]. These kinases are exploited by malignant cells and they contribute to the pathogenesis of several cancers, including ovarian cancer [22]. STAT3 is a latent transcription factor that is activated by upstream receptor kinases such as Janus activated kinases (JAKs) through cytokines such as interleukin-6 (IL-6), interleukin-10 (IL-10), granulocyte colony stimulating factor (G-CSF), leukaemia inhibitory factor (LIF) or leptin [23]. When these cytokines or growth factors bind to their respective receptors STAT3 is phosphorylated at Tyr-705, which leads to the formation of STAT3 homodimer that translocates to the nucleus where it binds to the promoter region of several genes including Mcl-1, survivin and cyclin-D1 [23]. STAT3 also activates vascular endothelial growth factor (VEGF) and is involved with the vascularisation of tumors [24].
Cisplatin is a common platinum-based drug used for the treatment of cancers, including ovarian cancer. It is a DNA strand cross-linking drug that generates DNA damage [25]. Most patients treated with cisplatin are either resistant to cisplatin or initially respond and then relapse [25]. Several mechanisms of resistance to platinum-based drugs have been described in cancers some of which include tolerance of the formation of platinum-DNA adducts, DNA repair mechanisms, altered cellular transport of the drugs, increased antioxidant production, and reduction of apoptosis [26–28]. In this context, altered gene expression affecting cellular transport, DNA repair, apoptosis and cell-cell adhesion have been observed in cisplatin resistant ovarian cancer patient samples [29–31]. In parallel, there are several DNA repair systems involved in the repair of cisplatin-DNA adducts [32] and activation of p53 and several other pathways have been shown to be involved [33], but the underlying mechanism(s) which specifically dictate cisplatin acquired resistance still remain unknown. In addition, cisplatin induced genotoxic stress has been shown to result in activation of multiple signal transduction pathways, among which are members of NFkappaB family [34] and mitogen activated protein kinase (MAPK) pathway family which includes the involvement of ERK, JNK and p38, the three major kinase cascades within the MAPK family [35]. Recently we have demonstrated that short-term single treatment of chemotherapy (paclitaxel or cisplatin) to ovarian cancer cell lines as well as isolated tumor cells from ascites results in the emergences of CSC-like cells which produces greater tumor burden in mice compared to untreated cells [10, 11, 36]. These results suggest that CSCs may have a crucial role in the emergence of aggressive tumors after chemotherapy treatment.
In this study, we present data that demonstrate that tumor cells isolated from the ascites of CR patients have enhanced JAK2/STAT3 pathway activation compared to isolated tumor cells from CN patients. We also demonstrate that a short-term single exposure of OVCA 433 and HEY ovarian cancer cell lines to cisplatin resulted in the activation of JAK2/STAT3 pathway in cisplatin surviving viable cells which coincided with the emergence of CSC-like cells. Both cisplatin-induced JAK2/STAT3 activation and CSC-like characteristics were inhibited by a low dose JAK2-specific small molecule inhibitor CYT387. The in vitro suppression of CSC-like characteristics and activation of the JAK2/STAT3 pathway by CYT387 was manifested in in vivo mouse xenografts with a reduced tumor burden. These data emphasize the need to explore further the effect of CYT387 in combination with chemotherapy in pre-clinical ovarian cancer models.
The human ovarian OVCA 433 and HEY cell lines were derived from the ascites and peritoneal deposit of a patient diagnosed with papillary cystadenocarcinoma of the ovary [10, 37]. The cell lines were grown as described previously [11].
Polyclonal antibody against phosphorylated (Tyr-705) STAT3 (P-STAT3), total STAT3 (T-STAT3), phosphorylated (Tyr-1007/1008) JAK2 (P-JAK2), total JAK2 (T-JAK2) and GAPDH were obtained from Cell Signalling Technology (Beverly, MA, USA). Antibodies against cytokeratin 7 (cyt7), Ki67, CA125, Oct4 and CD117 (c-Kit) used for immunohistochemistry were obtained from Ventana (Roche, Arizona, USA). CYT387 was obtained from Gilead Sciences (CA, USA).
Ascites was collected from patients diagnosed with Stages III–IV serous ovarian carcinoma and adenocarcinoma Not Otherwise Specified (NOS) (Table 1) after obtaining written informed consent under protocols approved by the Human Research and Ethics Committee (HREC approval # 09/09) of The Royal Women's Hospital, Melbourne, Australia. HREC approval #09/09 also obtained consent from participants to publish the results from this study provided anonymity of patients is maintained at all times.
| Sample | ID | Patient status | Stage | Grade | Treatment | Age at diagnosis | Overall Survival |
|---|---|---|---|---|---|---|---|
| Ascites | 77 | Chemoresistant (CR) | IV | High Grade Serous | Carboplatin and Paclitaxel 2 cycles | 53 years | 1 month as of 4/06/2013 |
| Ascites | 98 | Chemoresistant (CR) | IIIc | High Grade Serous | Paclitaxel 1 cycle | 59 years | 4 years 7 months |
| Carboplatin and Paclitaxel 6 cycles | |||||||
| OVAR16/VEG1106551 Trial 4 cycles | |||||||
| ICON62 Trial 7 cycles | |||||||
| Cisplatin 6 cycles | |||||||
| Doxorubicin Pegylated Liposomal 6 cycles | |||||||
| Docetaxel 3 cycles | |||||||
| REZOLVE3 Study 3 cycles | |||||||
| Ascites | 99 | Chemonaive (CN) | IIIc | High Grade Serous | No treatment | 64 years | NA |
| Ascites | 101 | Chemonaive (CN) | Unknown | Adenocarcinoma NOS | No treatment | 75 years | NA |
| Ascites | 102 | Chemoresistant (CR) | IV | High Grade Serous | Carboplatin and Paclitaxel 6 cycles | 50 years | 1 year 10 months as of 09/07/2014 |
| Carboplatin 3 cycles | |||||||
| Cisplatin 2 cycles | |||||||
| Doxorubicin Pegylated Liposomal (ongoing) | |||||||
| Ascites | 105 | Chemonaive (CN) | IV | High Grade Serous | No treatment | 83 years | 1 month as of 05/08/2014 |
For CN patients; ascites was collected after diagnosis, before treatment. CR patients; ascites was collected at recurrence after the patients had undergone the above described cycles of chemotherapy.
1OVCAR16/VEG110655: A phase III Study to evaluate the efficacy and safety of pazopanib monotherapy vs placebo in women who have not progressed after first line chemotherapy for epithelial ovarian, fallopian tube, or primary peritoneal cancer (Sponsor: GlaxoSmithKline). http://clinicaltrials.gov/ct2/show/NCT00866697.
2ICON6: A double-blind, placebo-controlled, three arm, randomised phase III trial of concurrent cediranib [AZD2171] (with platinum-based chemotherapy) and as a single agent maintenance therapy in women with ovarian cancer relapsing more than 6 months following completion of first line platinum-based treatment [Sponsor: Medical Research Council (UK)]. http://www.controlled-trials.com/ISRCTN68510403.
3REZOLVE: A phase II study to evaluate the safety and potential palliative benefit of intraperitoneal bevacizumab in patients with symptomatic ascites due to advance, chemotherapy-resistant ovarian cancer. http://www.anzgog.org.au/uploads/REZOLVE/2014%20Trials%20Summary%20REZOLVE.pdf.
The histopathological diagnosis, including tumor grades and stages was determined by independent staff pathologists as part of the clinical diagnosis (Table 1). Ascites was collected as they were received by the laboratory. For chemonaïve (CN) patients, ascites was collected at diagnosis before commencement of any treatment. Ascites was also collected from patients at the time of recurrence (CR). Patients in this group were not all treated identically and had previously received combinations of chemotherapies as described in Table 1. Ascites was collected from these patients at recurrence after the patients have completed combination chemotherapies described in Table 1.
Tumor cells from ascites were isolated as described previously [13]. Briefly, 500 ml of ascites was used to collect tumor cells. The ascites cells were seeded on low attachment plates (Corning Incorporated, NY) in MCDB:DMEM (50:50) growth medium supplemented with fetal bovine serum (10%), glutamine (2 mM) and penicillin/streptomycin (2 mM) (Life Technologies, CA, USA) after removal of the red blood cells by hypotonic shock using sterile MilliQ water as described previously [13]. Cells were maintained at 37°C in the presence of 5% CO2 and tumor cells floating as non-adherent population were collected after 2–3 days, and screened for CA125, EpCAM, cytokeratin 7 (CK7) and fibroblast surface protein (FSP) by Flow Cytometry to assess their purity [13]. Cells were passaged weekly and experiments were performed within 1–2 passages.
Ovarian cancer cell lines OVCA 433 and HEY were treated with cisplatin concentrations at which 50% growth inhibition was obtained (GI50 ∼ 5 μg/ml for OVCA 433 cells and 1 μg/ml for HEY cells for five and three days respectively) [11]. For CYT387 treatment, cells were screened for the response to different concentrations of CYT387 in both cell lines. The concentration of CYT387 that gave optimum inhibition of the active (phosphorylated) JAK2/STAT3 pathway in response to cisplatin in OVCA433 cells was ∼0.5 μM and 1 μM for HEY cells was used throughout the study. For combination treatment, OVCA 433 cells were treated with 5 μg/ml of cisplatin and with 0.125, 0.25 or 0.5 μM of CYT387 concentrations, while the HEY cells were treated with 1 μg/ml of cisplatin and 1 μM of CYT387. Treatment was performed for three-five days.
Immunofluorescence analysis of P-STAT3, T-STAT3, P-JAK2 and T-JAK2 was performed as described previously [11, 13, 36]. Images were captured by the photo multiplier tube (PMT) using the Leica TCS SP2 laser, and viewed on a HP workstation using the Leica microsystems TCS SP2 software. CellR software (Olympus Soft Imaging Solution) was used to perform semi-quantitative analysis amongst control and treated groups. For consistency and comparative results, as well as to avoid the edge effect, images were taken randomly at the centre of each well. CellR has inbuilt parameters which were set up with equal intensity for control and treated cells. These control measures were taken to justify the statistical differences between the control and treated preparations whilst avoiding biased measurement. To determine translocation of P-STAT3 from cytoplasm to nucleus in the absence and presence of treatments (cisplatin, CYT387 or combination of both), a semi-quantitative analysis was performed using CellR program. These measurements allowed quantification of P-STAT3 staining for a selection of cells which showed nuclear localisation in response to cisplatin, CYT387 and a combination of both.
Solid tumors derived from mice inoculated with HEY cells were homogenised using PowerLyzer™ 24 (MO BIO Laboratories Inc, Carlsbad CA, United States) according to manufacturer's instruction. RNA was extracted from the homogenized xenograft and cDNA synthesized as described previously [36]. Quantitative determination of mRNA levels of various genes was performed in triplicate using SYBR green (Applied Biosystems, Australia) as described previously [36]. The primers for Oct-4, CD117, and EpCAM have been described previously [36].
SDS-PAGE and Western blot was performed on cell lysates by the methods described previously [10]. Protein loading was monitored by stripping the membrane with Restore Western blot Stripping Buffer (Thermo Scientific, MA, USA) and re-probing the membrane with β-actin primary antibody (Sigma-Aldrich, Sydney, Australia).
Flow cytometry was used to assess the expression of cell surface markers and was performed as described previously [10–13]. 80–90% confluent cultures of OVCA 433 cells grown in the presence or absence of cisplatin for 5 days were collected and rinsed twice with PBS. 1 × 106 cells were incubated with primary antibody for 1 hour at 4°C and excess unbound antibody was removed by washing twice with PBS. Cells were stained with secondary antibody conjugated with phycoerythrin (PE) for 20 minutes at 4°C, washed twice with PBS and then re-suspended in 0.5 ml PBS prior to FACScan (Becton-Dickinson (Bedford, MA, USA) analysis. In each assay, background staining was detected using an antibody-specific IgG isotype. All data were analysed using Cell Quest software (Becton-Dickinson, USA). Results are expressed as mean intensity of fluorescence (MIF). Geo Mean value was used to calculate the relative expression of each cell surface marker analysed. IgG Geo mean was used as a negative control. For semi-quantitative analyses, expression of cell surface expression of CSC markers was calculated as a ratio between Geo mean of the cell surface marker over the Geo mean of the IgG.
3[[H]]-Thymidine uptake assay as a measure of cell proliferation was performed as described previously [13]. Briefly, 1 × 105 OVCA 433 or HEY cells untreated or treated with cisplatin or CYT387 + cisplatin were seeded in triplicate on 24 well plates. After 3 days, 0.5 μCi of [3[H]] thymidine was added to each well, and cells were incubated at 37°C for an additional 16 h. Cells were washed with PBS, harvested and lysed in 1% Triton and incorporation of [3[H]] thymidine was measured by liquid scintillation counting (Hidex 300SL, LKB Instruments, Australia).
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of the Laboratory Animals of the National Health and Medical Research Council of Australia. The experimental protocol was approved by the Ludwig Institute/Department of Surgery, Royal Melbourne Hospital and University of Melbourne's Animal Ethics Committee (Project-006/11), and was endorsed by the Research and Ethics Committee of Royal Women's Hospital Melbourne, Australia.
The animal experiments were performed as described previously [11, 13, 36]. The overall experiment was set out in six groups of mice which were as follows: (i) no treatment or control group, (ii) paclitaxel treatment group, (iii) CYT387 treatment group, (iv) paclitaxel+CYT387 treatments group, (v) cisplatin treatment group and (vi) cisplatin+CYT387 treatments group. The effects of paclitaxel treatment with or without CYT387 on HEY cell line in a mouse model has recently been published [36]. The current study focused on understanding the effects of cisplatin treatment in combination with CYT387 and as such included the overlapping control and CYT387 groups used in [36] in addition to cisplatin and cisplatin+CYT387 groups on HEY cells on a mouse model.
Briefly, female Balb/c nu/nu mice (age, 6–8 weeks) were obtained from the Animal Resources Centre, Western Australia. Animals were housed in a standard pathogen-free environment with access to food and water. HEY cells were treated with cisplatin (1 μg/ml) or CYT387 (1 μM) or cisplatin (1 μg/ml) plus CYT387 (1 μM) as described previously [36]. 5 × 106 cells surviving treatments after three days were injected intraperitoneally (ip) in nude mice. Mice were inspected weekly and tumor progression was monitored based on overall health and body weight until one of the pre-determined endpoints was reached. Endpoint criteria included loss of body weight exceeding 20% of initial body weight and general pattern of diminished well-being such as reduced movement and lethargy resulting from lack of interest in daily activities. Mice were euthanized and organs (liver, stomach, lungs, gastrointestinal tract, pancreas, uterus, skeletal muscle, colon, kidney, peritoneum, ovaries and spleen) and solid tumors were collected for further examination. Metastatic development was documented by a Royal Women's Hospital pathologist according to histological examination (H 8 E staining) of the organs.
For immunohistochemistry, formalin fixed, paraffin embedded 4 μm sections of the xenografts were stained using a Ventana Benchmark Immunostainer (Ventana Medical Systems, Inc, Arizona, USA) as described previously [11]. Immunohistochemistry images were taken using Axioskop 2 microscope, captured using a Nikon DXM1200C digital camera and processed using NIS-Elements F3.0 software. Images were scored independently by four reviewers blind to the molecular data as previously described [11, 36, 38].
Data are presented as mean ± SEM. Treatment groups were compared with the control group using one way- ANOVA and Dunnett's Multiple Comparison post-tests. A probability level of p < 0.05 was adopted throughout to determine statistical significance.
The expression of phospho (P)-JAK2 and P-STAT3 in isolated tumor cells from the ascites of CN and CR patients were analysed by immunofluorescence staining. In the three ascites samples analyzed from each CN and CR groups, the expression of P-JAK2 and P-STAT3 were significantly higher in CR group compared to CN group (Fig. 1). The expressions of P-JAK2 and P-STAT3 were standardized to total (T)-JAK2 and T-STAT3 in each case.
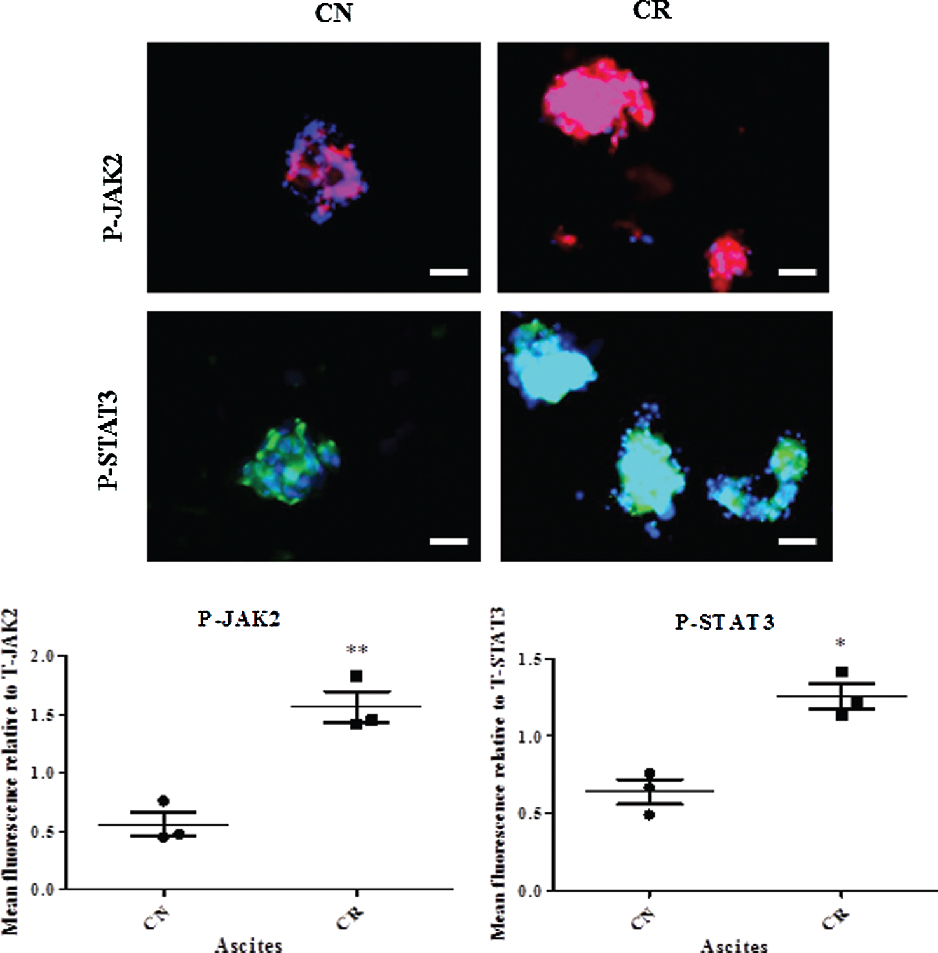 |
Figure 1. Enhanced expression of P-JAK2 and P-STAT3 in isolated tumor cells obtained from the ascites of CR patients compared to CN patients. Expressions of P-JAK2 and P-STAT3 in isolated tumor cells from ascites of CN and CR patients were analysed by immunofluorescence using rabbit polyclonal (red) and mouse monoclonal (green) antibodies as described in the Materials and Methods. Cellular staining was visualized using secondary Alexa 488 (green) or Alexa 590 (red) fluorescent labelled antibodies. Nuclear staining was visualized using DAPI (blue) staining. Images are representative of three independent patients in each group. The mean fluorescence intensity of P-JAK2 and P-STAT3 were quantified using Cell-R software (Olympus Soft Imaging Solutions) and standardised to T-JAK2 and T-STAT3. Magnification 200×; scale bar = 50 μM. |
We assessed the levels of P-JAK2 and P-STAT3 in response to cisplatin treatment in viable OVCA 433 cells by Western blot (Fig. 2A). The expression of P-JAK2 was increased as a result of cisplatin treatment (5 μg/ml) after 5 days. Cisplatin induced phosphorylation of JAK2 was inhibited by the addition of CYT387 at 0.5 μM concentration, while differential effects were observed at 0.125 and 0.25 μM concentrations. The expression of total JAK2, on the other hand, under these conditions was evenly expressed across the samples. Equal protein loading was determined by the expression of β-actin (Fig. 2A). The activation of P-JAK2 coincided with the downstream activation of STAT3 in response to cisplatin treatment (Fig. 2B); however, inhibition of cisplatin-induced P-STAT3 was observed at all CYT387 concentrations used (Fig. 2B). β-actin confirmed the equal loading of protein in each sample (Figs. 2A and 2B).
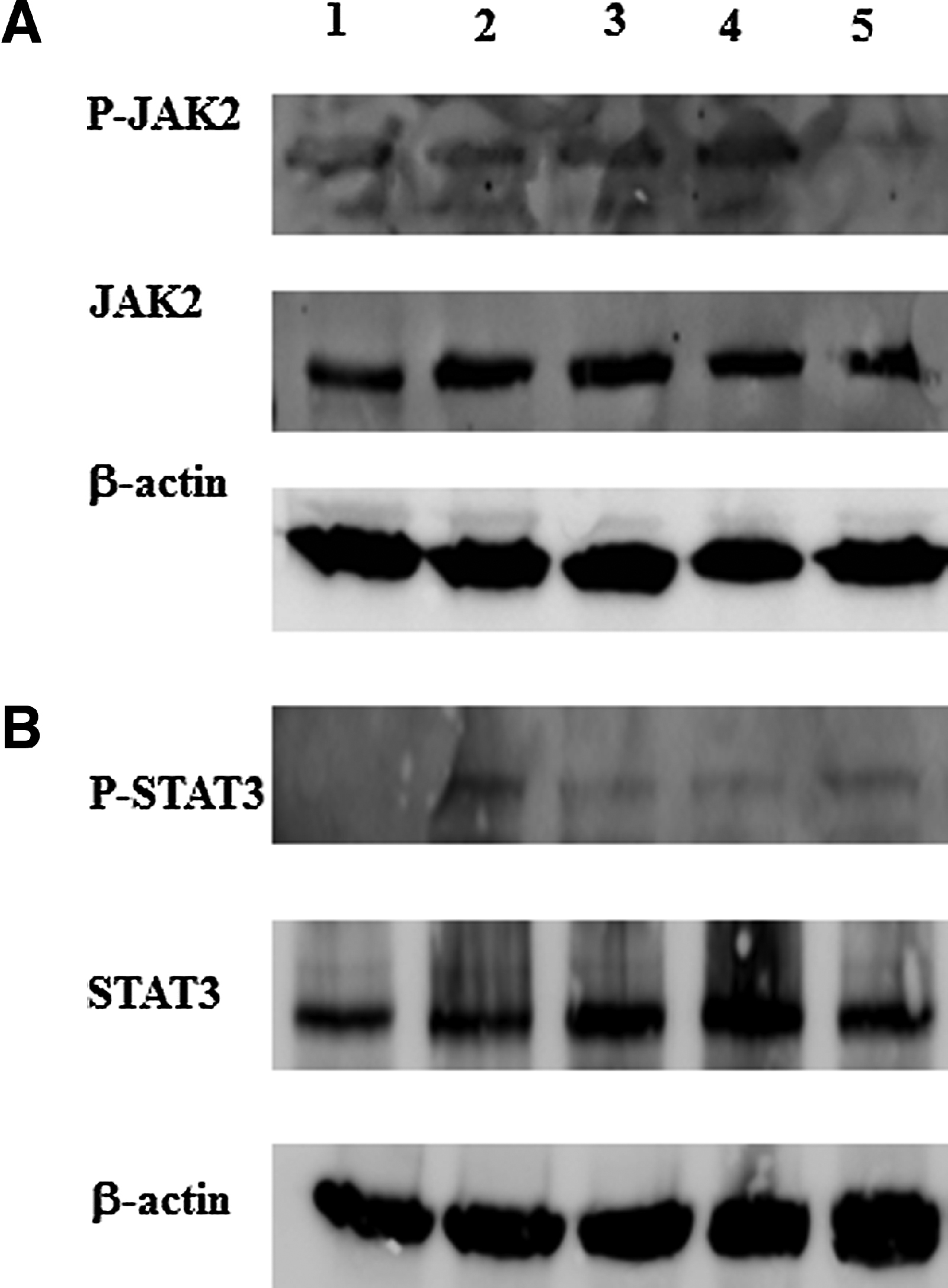 |
Figure 2. Activation of JAK2 and STAT3 in response to cisplatin treatment and inhibition by CYT387 in OVCA 433 cells. OVCA 433 cells were treated with cisplatin and a combination of cisplatin and different concentrations of CYT387. Cell lysates were prepared as described in the Methods and Material section and assessed by Western blot for the activation of (A) JAK2 and (B) STAT3. Samples are arranged as follows: 1) control, 2) cisplatin (5 μg/ml), 3) cisplatin (5 μg/ml) + CYT387 (0.125 μM), 4) cisplatin (5 μg/ml) + CYT387 (0.250 μM), 5) cisplatin (5 μg/ml) + CYT387 (0.5 μM). Total protein loading was determined by probing the membranes for β-actin. Results are representative of two independent experiments. |
We next assessed the activation of P-JAK2 and STAT3 by cisplatin and its concomitant inhibition by CYT387 by immunofluorescence (Fig. 3A). In previous experiments with Western blot we were able to show that CYT387 at a concentration of 0.5 μM inhibited cisplatin-induced JAK2/STAT3 activations. Thus, for this series of experiments CYT387 at 0.5 μM concentration was used. To determine whether treatment with cisplatin and CYT387 alone or in combination had a significant impact on the translocation of P-STAT3 from cytoplasm to nucleus, a semi-quantitative immunofluorescence analysis was performed. The expression of P-STAT3 in the control cells was mostly confined to the cytoplasm while very few nuclear staining was evident in some sections (Fig. 3A). However, P-STAT3 was predominantly translocated to the nucleus in OVCA 433 cells treated with cisplatin, yet some staining in the cytoplasm was also observed. In addition to the activation of P-STAT3, cisplatin also induced EMT in cisplatin treated OVCA 433 cells as we have reported previously [10] (Fig. 3A). A comparison between the control and cisplatin treated samples revealed that the translocation of P-STAT3 to the nucleus in response to cisplatin treatment was significantly increased (p < 0.05) (Fig. 3B). However, this translocation was decreased when CYT387 was added concomitantly in addition to cisplatin (∼2-fold, p < 0.05) (Fig. 3B). On the other hand, treatment with CYT387 on its own had no significant effect on the translocation of P-STAT3 to the nucleus compared to the control untreated cells (Fig. 3B).
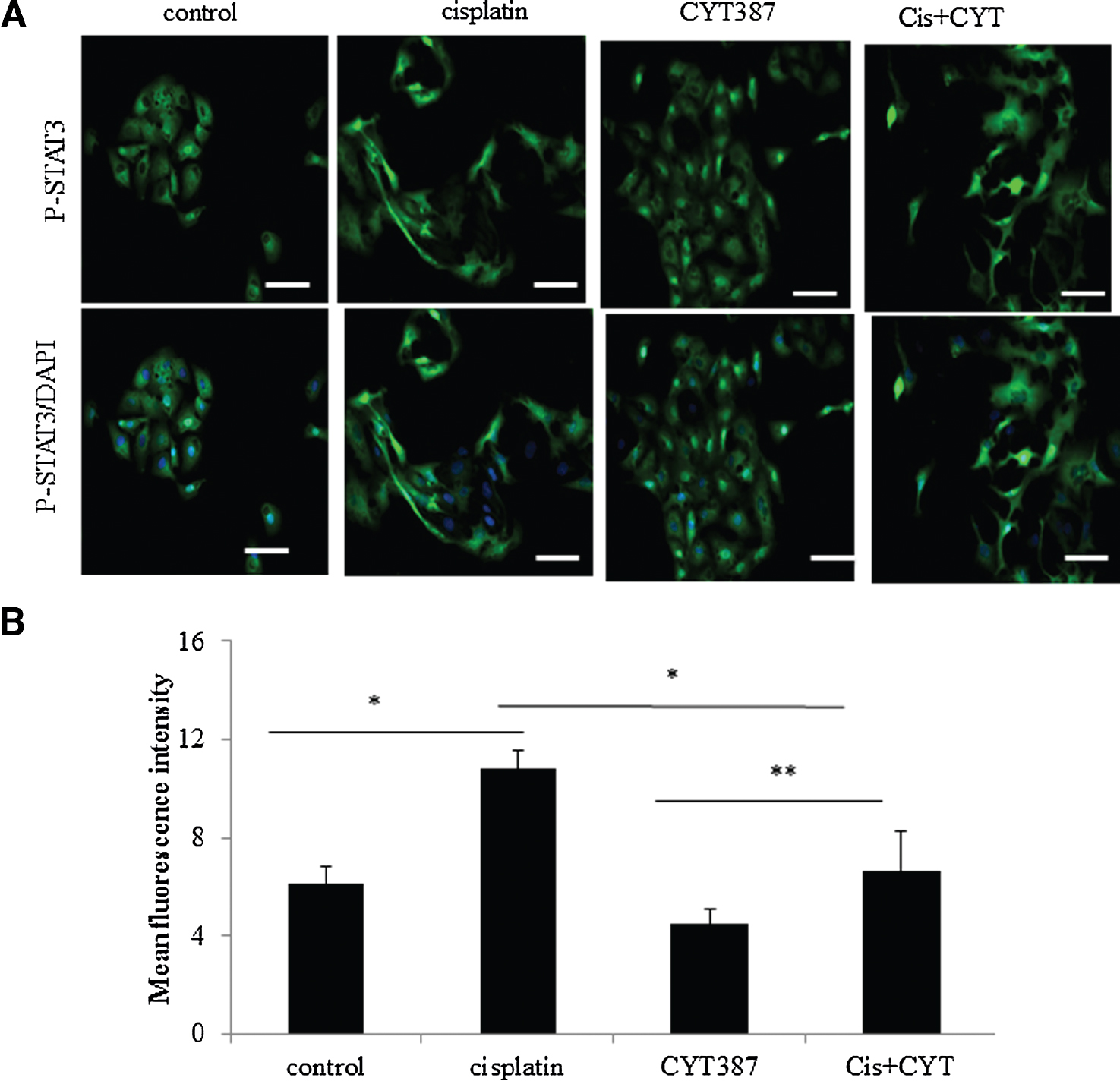 |
Figure 3. Immunofluorescence evaluation of P-STAT3 in OVCA 433 cell line treated with cisplatin and CYT387. (A) Immunofluorescence evaluation of P-STAT3 was performed as described in Figure 1. Activation of P-STAT3 was detected by a mouse monoclonal antibody and staining was visualized using the secondary Alexa 488 (green) fluorescent-labelled antibody. The nuclei were detected by DAPI (blue) staining. Results are representative of five independent experiments. Magnification was 200X; scale bar = 10 μm. (B) Mean fluorescence intensity of P-STAT3 stained cells translocated to the nucleus in the control, cisplatin (5 μg/ml), CYT387 (0.5 μM) and combination of cisplatin (5 μg/ml) + CYT387 (0.5 μM) (Cis+CYT) treatment of OVCA 433 cells was evaluated using Cell-R software (Olympus Soft Imaging Solutions). Results are expressed as the average intensity of five independent samples +SEM. Significance, * p < 0.05, **p < 0.01. |
Consistent with OVCA 433 cells, treatment with cisplatin resulted in the activation of the JAK2/STAT3 pathway in the ovarian cancer HEY cell line (Supplementary Fig. 1). This observation was confirmed by immunofluorescence which demonstrated significant enhancement in the level of phosphorylated JAK2 (Tyr-1007/1008) and downstream STAT3 (Tyr-705) compared to control untreated cells (Supplementary Fig. 1). Both P-JAK2 and P-STAT3 in cisplatin-treated cells were found to be localized in the nucleus as well as cytoplasm of the cisplatin-treated cells (Supplementary Fig. 1). The expression of T-JAK2 and T-STAT3 which was localized mostly in the cytoplasm under the same experimental conditions remained unchanged (Supplementary Fig. 2).
We have previously shown the enhancement of CSC-like phenotypes in OVCA 433 and HEY ovarian cancer cell lines in response to short-term treatment with cisplatin [11]. In this study we demonstrate the suppression of expression of cisplatin-induced CSC-like markers by CYT387 in OVCA 433 cells. The cell surface expression of CSC markers in control untreated and cisplatin treated OVCA 433 was assessed by Flow cytometry. Low or no expression of CD117 and CD133 was observed in control cells. In contrast, high expression of CD44 and EpCAM was evident in control cells. The expression of CD117, CD133, CD44 and EpCAM were all enhanced with cisplatin treatment, while no change in the expression of these markers were observed between control and CYT387 treatment with the exception of CD44 expression where it was observed to be decreased (Figs. 4A and 4B). Importantly, no visual reduction in the expression of CSC markers when cisplatin and CYT387 (0.5 μM) were used concomitantly was observed compared to only cisplatin treated cells (Fig. 4B).
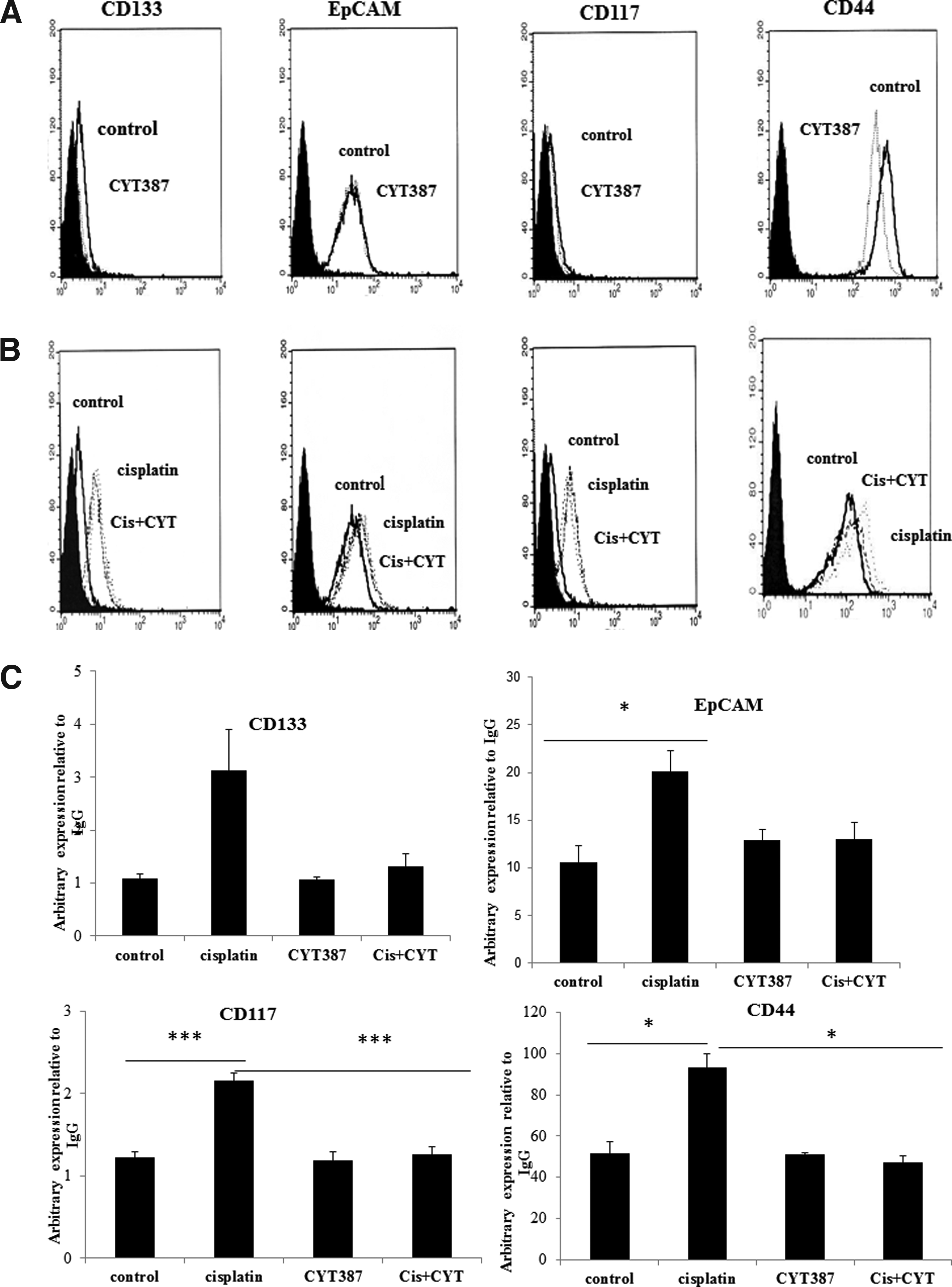 |
Figure 4. Flow cytometry assessment of CSC markers expressed on OVCA 433 cells in response to cisplatin and CYT387 treatments. Flow cytometry assessment of CSC markers in control, cisplatin (5 μg/ml), CYT387 (0.5 μM) and combination of cisplatin (5 μg/ml) + CYT387 (0.5 μM) (Cis+CYT) was performed as described in Methods and Materials. (A) The filled histogram in each figure represents control IgG, the dotted line, in panel A, represent the antigen expression of cells treated with CYT387 (0.5 μM). (B) The filled histogram in each figure represents control IgG, the dark dotted lines represent CSC expression in response to cisplatin (5 μg/ml) while light dotted lines represent expression of CSCs in response to Cis+CYT. Results are representative of three independent experiments and represented by arbitrary fluorescence. (C) Flow cytometry semi-quantitative analysis of the expression of CSC markers was performed as described in Methods and Materials. Results are expressed as the difference between the arbitrary expression of CSC markers to that of IgG ± SEM of three independent samples. Significance, *p < 0.05 and ***p < 0.001. |
To evaluate further the effect of CYT387 in combination with cisplatin on the expression of CSC markers, a semi-quantitative analysis was performed which took into account the expression of CSC markers by Flow cytometry and standardised that to respective IgG controls as discussed in the Methods and Materials. The expression of CD133 in control, CYT387 treatment only and a combination of cisplatin and CYT387 treatments remained the same (Fig. 4C). Despite an approximate three-fold increase in CD133 expression in cisplatin treated OVCA 433 cells compared to control untreated cells, no significant difference was observed between the two groups (p > 0.054) (Fig. 4C). There was a decrease in the expression of CD133 in the presence of both cisplatin and CYT387 compared to cisplatin treatment only but it was not significant. The expression of EpCAM, however, was significantly enhanced in cisplatin-treated OVCA 433 cells compared to control cells (Fig. 4C). However, no significant change between the cisplatin treatment and cisplatin +CYT387 treatments were observed (Fig. 4C). The expression of CD117 was significantly enhanced in the cisplatin treated cells compared to control cells (p < 0.001) (Fig. 4C). This enhancement in cisplatin-induced expression was significantly reduced by the addition of CYT387 in the presence of cisplatin (p < 0.001). Similar results were observed for CD44 expression which was significantly enhanced by cisplatin treatment compared to control cells (p < 0.01) and significantly reduced by the addition of CYT387 in combination with cisplatin (p < 0.01) (Fig. 4C).
The effect of CYT387 (0.5 μM) to inhibit the growth of OVCA 433 cells in presence of cisplatin was assessed by the [3[H]]-thymidine uptake assay. Cisplatin treatment of OVCA 433 cells significantly reduced cell proliferation by approximately 54% when compared to control (p < 0.05) (Fig. 5). Even though the combination of cisplatin and CYT387 was effective in reducing the cell proliferation by a further 20% it was not significant compared to cisplatin alone treatment (Fig. 5). However, a comparison between CYT387 and combination of CYT387 and cisplatin treatments resulted in a significant reduction in proliferation (p < 0.05) (Fig. 5). CYT387 on its own had no significant effect on the proliferation of OVCA 433 cells compared to control cells (Fig. 5).
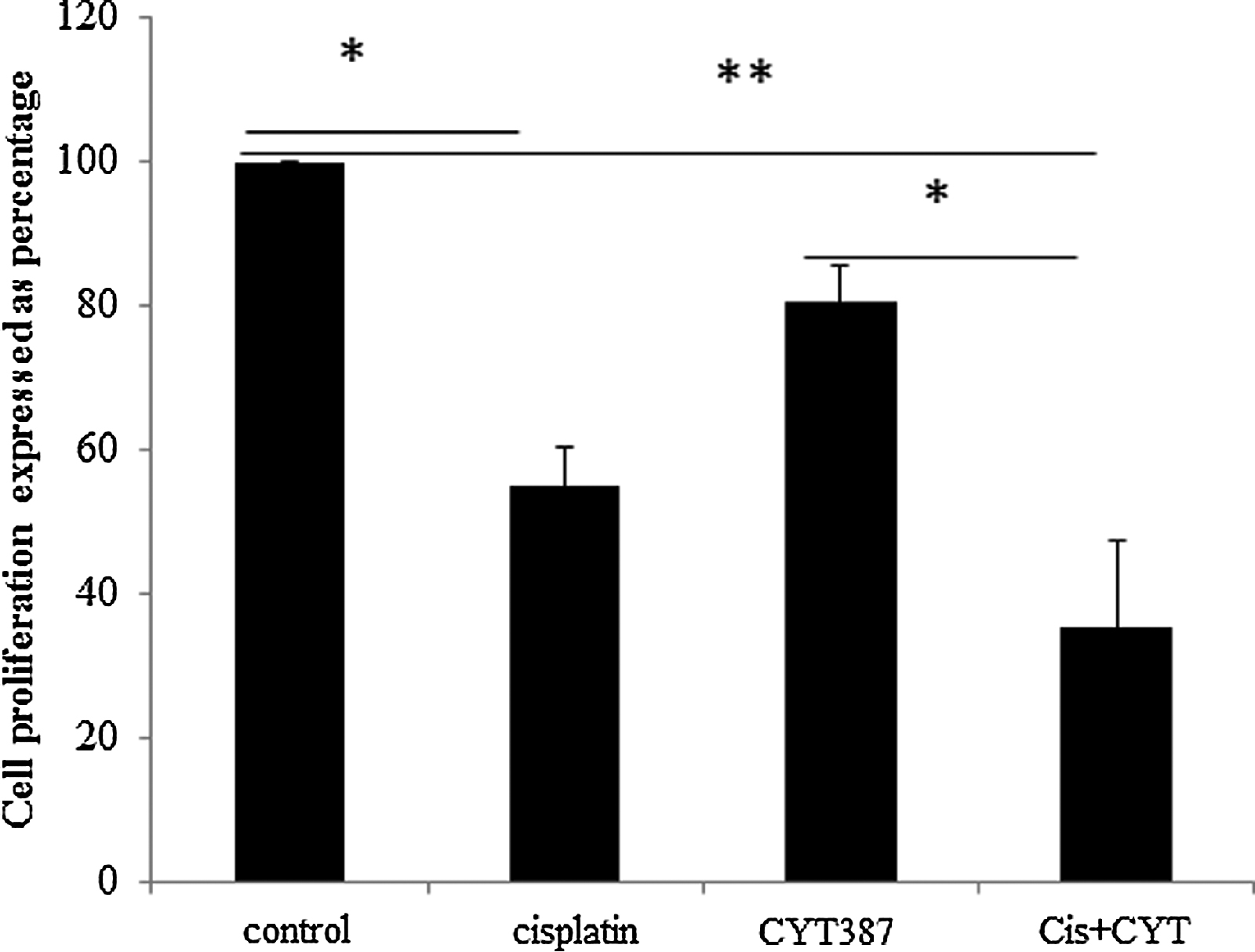 |
Figure 5. Effect of CYT387 on the proliferation of OVCA 433 cells. OVCA 433 cells were treated with cisplatin (5 μg/ml) or CYT387 (0.5 μM) or cisplatin (5 μg/ml) + CYT387 (Cis +CYT) (0.5 μM) for three days. [3[H]]-thymidine was added and the cells were harvested as described in the Methods and Materials. The data is a representation of three independent experiments performed in triplicate. Significant variations between the groups are indicated by *P < 0.05, ** P < 0.01. |
Human ovarian OVCA 433 cells could not be grown in nude mouse intraperitoneally, as observed previously [39]. The effect of the addition of CYT387 in conjunction with cisplatin treatment was tested in in vivo mouse intraperitoneal (ip) HEY xenograft model used previously [36]. Mice (n = 5) injected with untreated HEY cells developed solid tumors in the form of 3–4 small lesions (<0.5 cm3) in the peritoneum within six weeks. The average weight of the debulked tumors from the five control mice injected with untreated HEY cells weighed approximately 4.8% ± 2.3 of the total body weight (Fig. 6). In contrast, mice injected with the same number (5 × 106) of cisplatin-surviving HEY cells produced a significantly larger tumor burden within the same time period, with the average tumors weighing ∼ 11% ± 3 of the total body weight (Fig. 6). On the other hand, tumors in mice injected with CYT387 plus cisplatin treated cells weighed on average 3.2% ± 2 of the total mouse body weight (Fig. 6). The average tumor weight in mice injected with CYT387 only treated HEY cells was ∼ 3.5% ± 1.3 of the total body weight. In short, no significant difference in the tumor burden was observed between groups of mice injected with control untreated HEY cells or HEY cells treated with CYT387 (Fig. 6). On the other hand, significantly lower tumor burden was observed in mice injected with HEY cells treated with a combination of cisplatin and CYT387 versus mice injected with cells treated with cisplatin alone (Fig. 6). These results suggest that CYT387 in combination with cisplatin reduces the tumor burden induced by cisplatin only treatment however, CYT387 on its own had no significant effect in reducing the tumor burden compared to control HEY cells-derived tumor burden.
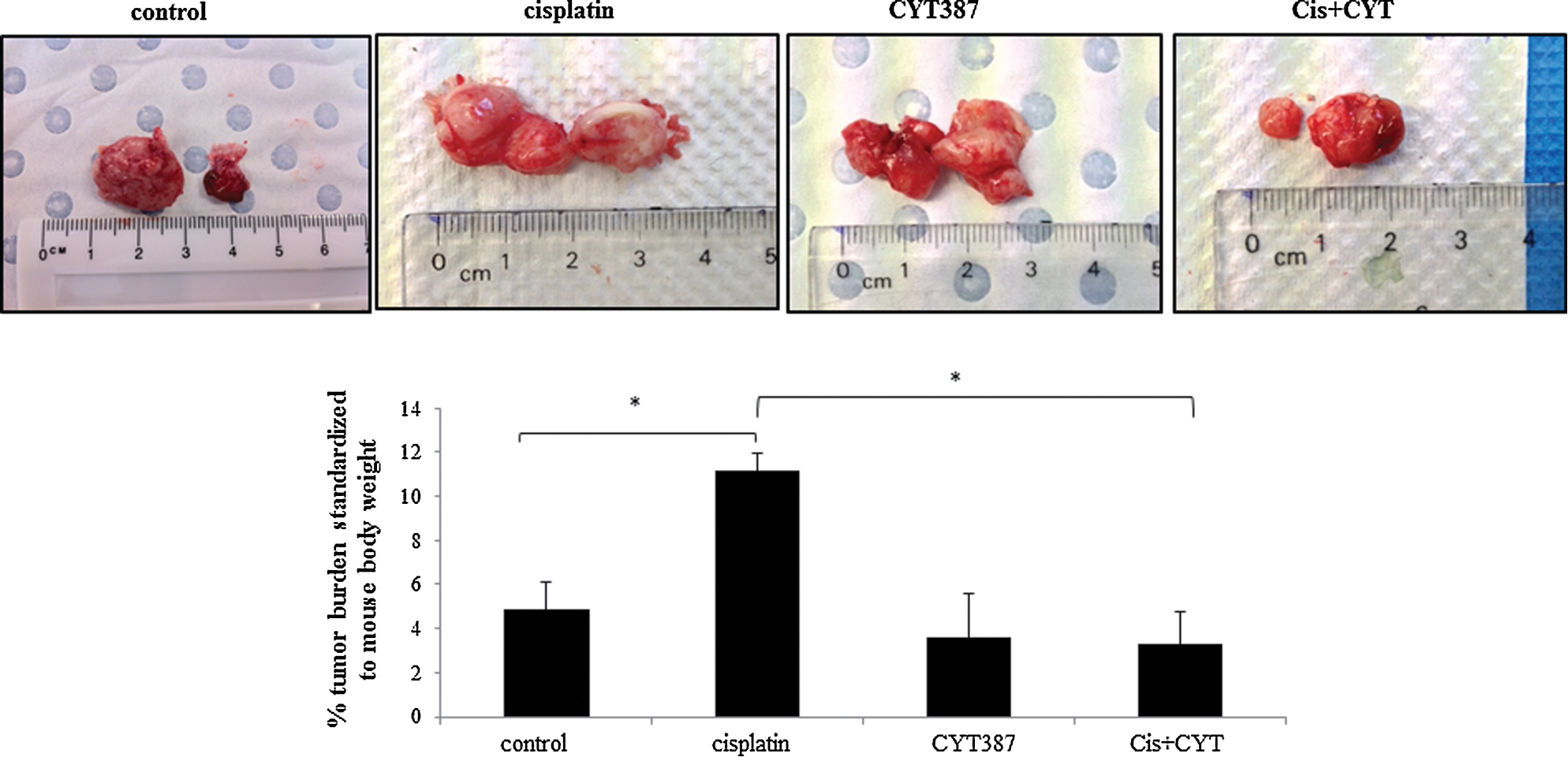 |
Figure 6. Tumor burden in mice injected with control or cisplatin or CYT387 or a combination of cisplatin plus CYT387-treated cells. Total tumor burden obtained from mice 6 weeks after ip injection of control, cisplatin-treated, cisplatin plus CYT387-treated and combination of both CYT387 and cisplatin-treated (Cis+CYT) HEY cells (n = 5/group). 5 × 106 cells were inoculated in each case. *P < 0.05, significant increase in tumor burden in cisplatin-treated HEY cells derived tumors compared to control untreated group; and cisplatin-treated HEY cells derived tumors to Cis+CYT-treated cells derived tumors. Images represent total tumors debulked from one mouse in each group. Control and CYT387 groups are the same as described in [36]. |
Debulked mouse tumors from mice inoculated with control, cisplatin, CYT387 or cisplatin plus CYT387 treated HEY cells were analysed using immunohistochemistry. We performed immunohistochemistry analysis of the active (phosphorylated) and total JAK2 and STAT3 levels in mouse xenografts in all four groups. Cisplatin-treatment cell derived tumors displayed significantly enhanced staining for both P-JAK2 and P-STAT3 compared to tumors derived from untreated or CYT387-treated HEY cells derived tumors (Fig. 7A–C). On the other hand, tumors derived from HEY cells treated with cisplatin plus CYT387 displayed significantly decreased staining for P-JAK2 and P-STAT3 compared to tumors derived from cisplatin-treated cells, but expressed P-JAK2 and P-STAT3 at the same level as tumors derived from control untreated or CYT387-treated HEY cells (Fig. 7A–C). The expression of T-JAK2 and T-STAT3 remained unchanged in control and treatment groups. Coinciding with the activation of the JAK2/STAT3 pathway, immunohistochemistry analysis of mouse tumors for the CSC marker CD117 (c-kit), the embryonic stem cell marker Oct4 and the ovarian cancer marker CA125 revealed significantly enhanced staining in xenografts derived from cells surviving cisplatin treatment compared to control untreated cells (Fig. 8A-B). The expression of CD117, Oct4 and CA125 were reduced significantly in CYT387 plus cisplatin treated cells-derived xenografts compared to cisplatin only treated cells-derived xenografts, and was more comparable to xenografts derived from control untreated or CYT387-treated cells xenografts (Fig. 8A-B). To determine if the changes in CSC markers seen in mouse xenografts derived from cisplatin-treated and cisplatin plus CYT387-treated HEY cells were consistent at the mRNA level; q-PCR was performed on cDNA prepared from RNA extracted from these tumors. Compared to xenografts derived from control untreated HEY cells, tumors derived from cisplatin surviving cells showed significant enhancement of mRNA expression of CD117 and EpCAM (Fig. 9). Although cisplatin treatment derived tumors showed significant enhancement of Oct4 at the protein level, and an apparent increase at the mRNA level, this enhancement was not statistically significant. CYT387 on its own had no significant effect on the mRNA expression of CD117, EpCAM and Oct4 compared to control cells. However, concomitant addition of cisplatin and CYT387 addition to cells led to significant reduction in the mRNA expression of CD117 and EpCAM expression compared to cisplatin only effect (Fig. 9). The mRNA expression of Oct4 when cisplatin and CYT387 were added together compared to cisplatin only treatment showed no significant difference (Fig. 9).
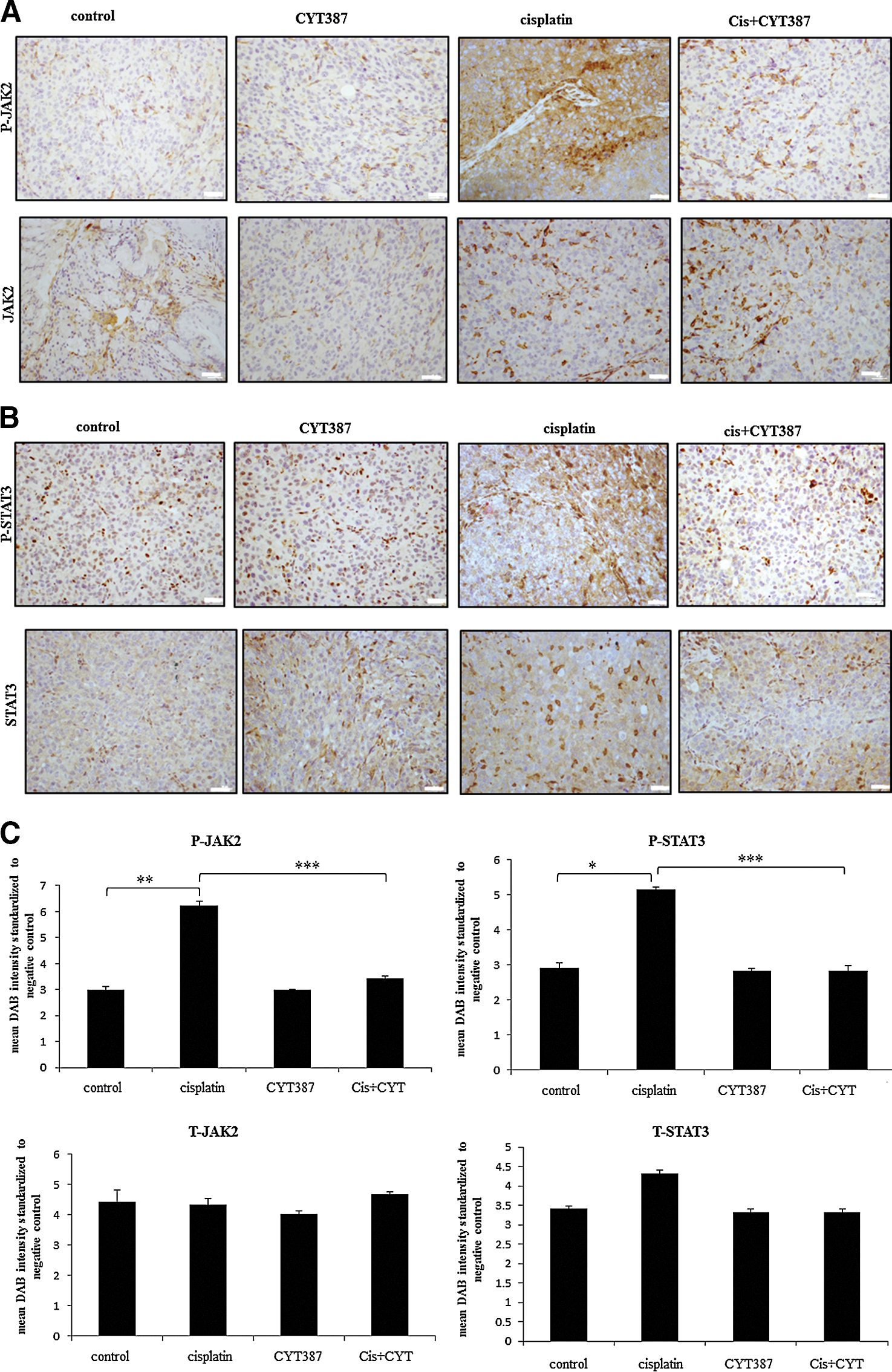 |
Figure 7. Immunohistochemical expression of P-JAK2, T-JAK2, P-STAT3 and T-STAT3 in mouse tumors generated from ip transplantation of control or cisplatin, CYT387 and a combination of cisplatin plus CYT387 treated HEY cells. Tumor sections were stained for the expression of (A) P-JAK2, T-JAK2, (B) P-STAT3 and T-STAT3 as described in Materials and Methods. (C) The data was obtained from three independent xenografts and scored by four blind reviewers as described in Materials and Methods. Significant variations between the groups are indicated by *P < 0.05, **P < 0.01 and ***P < 0.001. Magnification 200X, scale bar = 10 μm. |
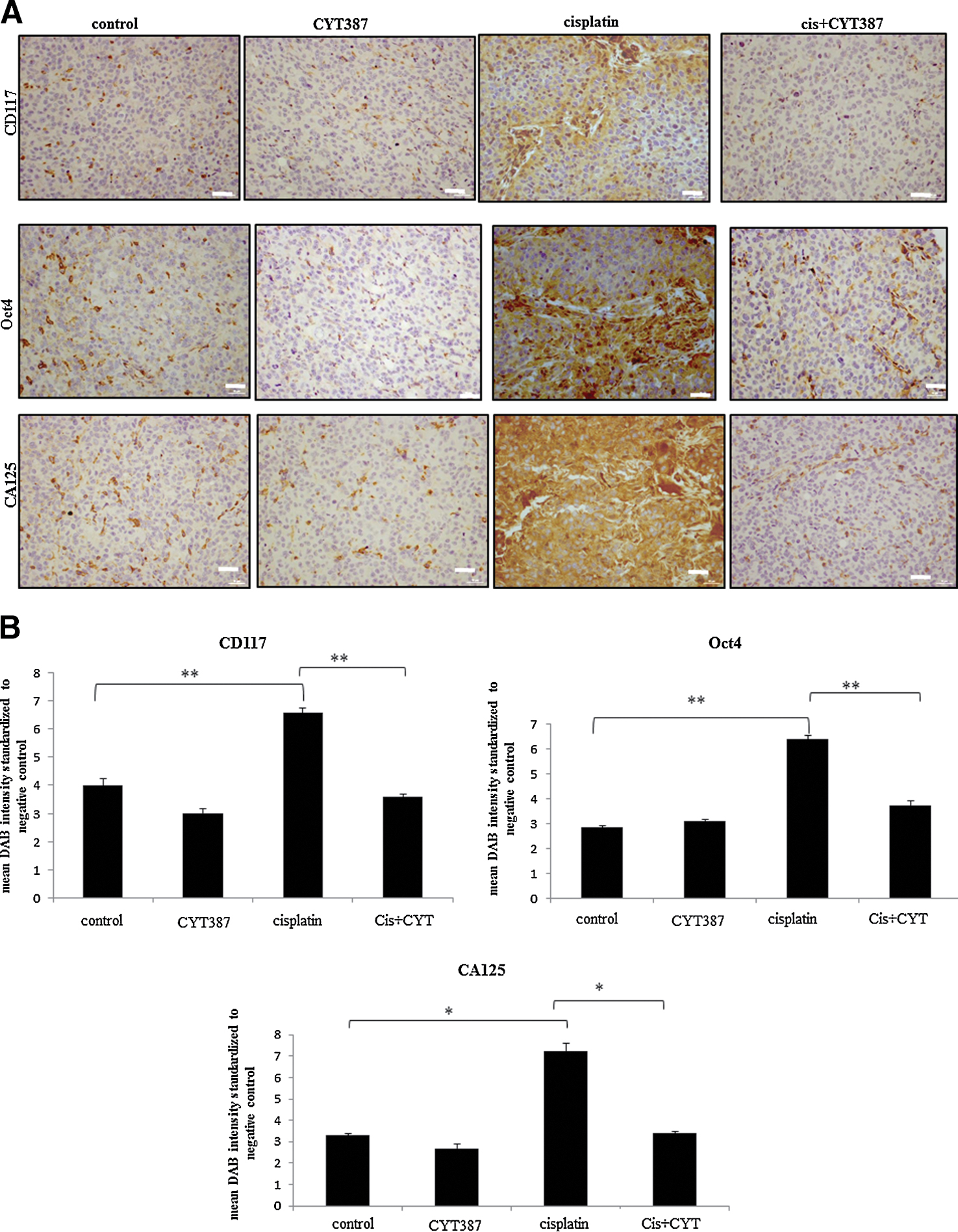 |
Figure 8. Expression of CSCs and CA125 in mouse tumors generated from ip transplantation of control or cisplatin or CYT387 and a combination of cisplatin and CYT387-treated HEY cells by immunohistochemistry. (A) Tumor sections were stained and (B) scoring for the expression of Oct4, CD117 and CA125 was performed as described in Figure 8. Significant variations between the groups are indicated by *P < 0.05 and **P < 0.01. Magnification 200X, scale bar = 10 μm. |
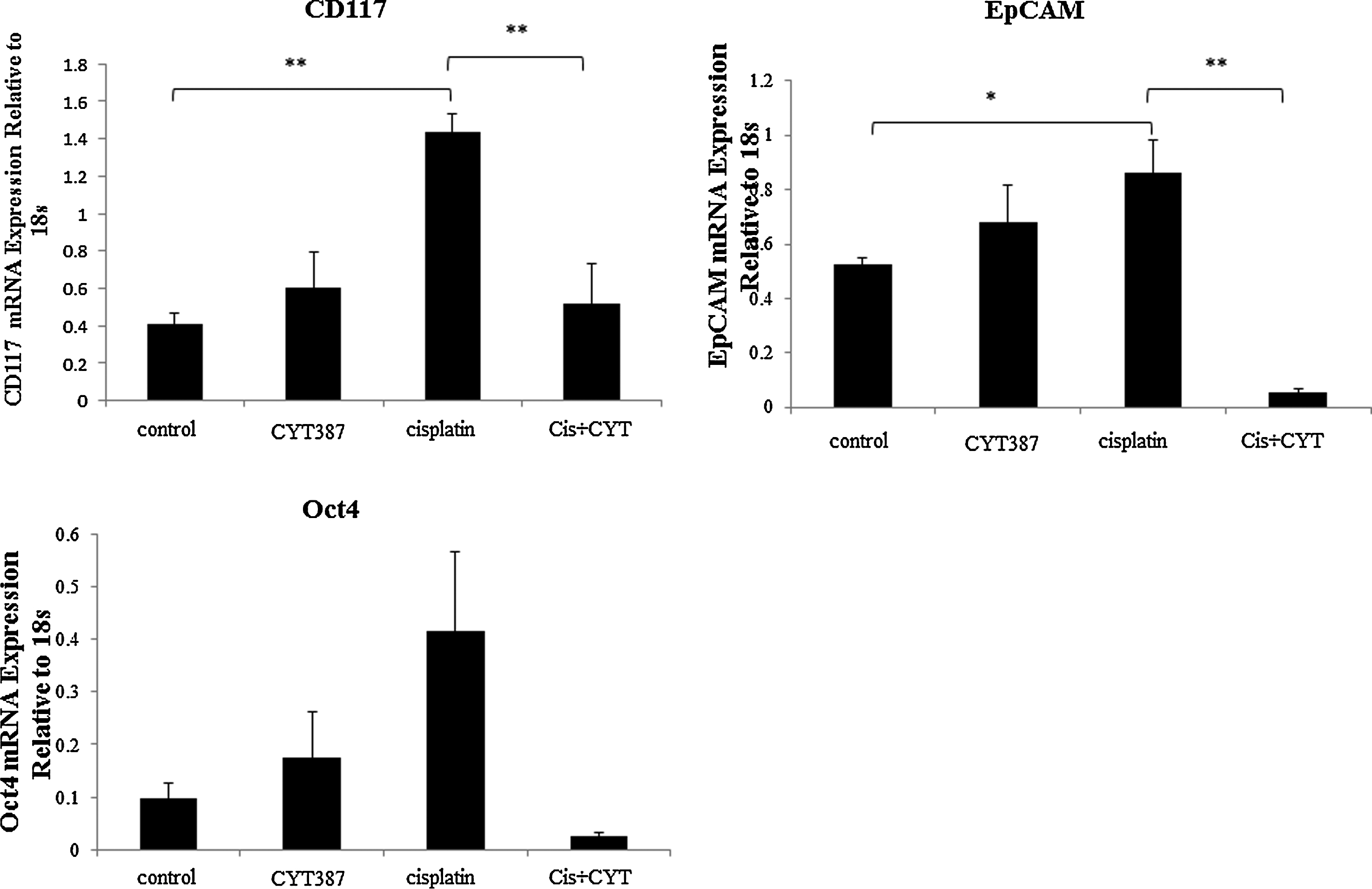 |
Figure 9. The mRNA expression of CSC markers in control or cisplatin or CYT387 or cisplatin plus CYT387-treated HEY cells-derived xenografts. RNA from control and treated HEY cells-derived xenografts was extracted, cDNA was prepared and q-PCR for EpCAM, CD117 and Oct4 was performed as described in the Materials and Methods section. The resultant mRNA levels were normalized to 18S mRNA. The experiments were performed using four independent samples in triplicate. Significant variations between the groups are indicated by *P < 0.05 and **P < 0.01. |
Negative IgG controls for Figures 7A–B and 8A are provided in Supplementary Figure 3.
The tumor infiltration pattern within the peritoneal cavity in response to cisplatin, CYT387, cisplatin plus CYT387 was assessed using H8E staining. In line with our previous study [11, 36], sections of mouse organs (pancreas, liver, intestine, colon, and kidney) displayed infiltrating tumors with epithelial morphology. Although the addition of CYT387 with cisplatin resulted in the significant reduction of tumor burden, an observation using a minimum of three mice in all treatment groups revealed a similar invasion pattern in all treatment regimens (images for pancreas and liver presented in Supplementary Fig. 4).
Ovarian cancer cells with CSC-like features have been proposed to be resistant to cancer therapies [9–12], suggesting that their effective elimination may require the identification of signaling pathways on which they are dependent for their survival. In this study we demonstrate enhanced activation of STAT3 in tumor cells isolated from the ascites of CR patients compared to tumor cells derived from the ascites of CN patients. We also demonstrate activation of the JAK2/STAT3 pathway in cisplatin-treatment surviving viable ovarian cancer cells in vitro with concurrent enhancement of CSC-like traits. In addition, suppression of this cisplatin treatment-induced activation of JAK2/STAT3 pathway, by a potent JAK2-specific inhibitor, CYT387, not only resulted in the abrogation of CSCs in vitro but also in vivo and correlated with the reduction of a significant tumor burden in mice. These data suggest that combination of CYT387 with cisplatin could be a therapeutic option for chemoresistant ovarian cancer patients as it targets the JAK2/STAT3 pathway which is required for the CSC-like chemotherapy resistant cells.
The presence of ascites has been associated with a poor prognosis [5, 40]. However, the origin and phenotype of the cellular content of ascites and its association with chemoresistance and recurrence is poorly understood. Microscopic inspection of ascites usually display a complex heterogeneous image which consists of single cells and multicellular aggregates or spheroids and non-cancer inflammatory single cells, cancer-associated fibroblasts, myeloid cells and activated mesothelial cells [40]. Recently, we have developed a novel method of isolating tumor cells from the ascites of ovarian cancer patients [13]. Using this method, we have previously shown that isolated tumor cells from the ascites of CR patients demonstrate CSC-like characteristics compared to tumor cells isolated from CN patients [13]. In this study, we demonstrate that ascites-derived tumor cells from CR patients have an enhanced level of activated JAK2/STAT3 pathway compared to tumor cells of CN patients. These observations are consistent with one previous study where metastatic and drug resistant recurrent ovarian tumors showed a significantly higher IL6 induced STAT3 phosphorylation compared to the matched primary tumors [41]. Furthermore, a monoclonal anti-IL6 antibody, siltuximab (CNTO 328) was shown to suppress IL-6 induced STAT3 phosphorylation and nuclear translocation and decrease the expression of STAT3 downstream targets and sensitize paclitaxel resistant ovarian cancer cell lines to chemotherapy [41].
CYT387 is an orally available, bioactive inhibitor of the JAK1/2 pathway currently in Phase III clinical trial for the treatment of myelofibrosis, a frequently diagnosed fatal myeloproliferative disorder [42]. The preliminary data on the drug has shown durable anemia responses and favorable toxicity profile in patients, suggesting CYT387 to be the best candidate among the JAK inhibitors so far discovered for the management of myelofibrosis. CYT387 has shown effectiveness in a JAK2V617F mutation-associated animal model where it inhibited STAT3 functions associated with constitutively activated JAK2, by normalising inflammatory cytokines [43]. Preclinical analysis has shown CYT387 to be well tolerated in mice when administered orally at doses up to 50 mg/kg of body weight, with no sign of overt toxicity [43]. Besides the myeloproliferative disorder, CYT387 has also potential for the treatment of phenotypically diverse myelomas and recently has shown efficacy in K-RAS driven lung cancer model by inhibiting the innate immune signaling kinase Tank-binding kinase-1 [44, 45]. In this study we demonstrate another novel cellular effect of CYT387 in vitro and in a in vivo mouse model where it suppressed the activated STAT3 in response to cisplatin-treatment in vitro, and produced significantly smaller tumors when injected in mice with ovarian cancer cells treated in vitro with cisplatin and CYT387 compared to the tumors derived from mice injected with cisplatin only treated cells. These results correlated with the abrogation of CSC-like characteristics in vitro and in vivo mouse xenografts when CYT387 was added in conjunction with cisplatin compared to cisplatin only treatment.
The current study is an extension of our recently published study where we have shown similar effects of CYT387 in combination with paclitaxel [36]. Hence, one can say that even though the mode of action of cisplatin and paclitaxel are considerably different, both drugs are able to activate the JAK2/STAT3 pathway in surviving ovarian cancer cells which tolerated the cytotoxic effects of these chemotherapies. Platinum-based cisplatin or carboplatin in combination with taxane-based paclitaxel are the most frequently used chemotherapeutic drugs for the treatment of ovarian cancer for the last thirty years [4]. Considering that the overall survival of ovarian cancer patients has not improved significantly for the last thirty years is related to the observation that certain population of ovarian cancer cells exhibit resistance to these chemotherapies and are able to regrow and regenerate tumors locally and/or at distant sites after chemotherapy treatments [10, 46]. In this study, we propose a putative explanation for cisplatin and paclitaxel resistance relating to the CSC hypothesis and activation of JAK2/STAT3 pathway as a mechanistic pathway for the generation of residual CSC-like cells. Using two very different ovarian cancer cell lines originating from ascites (OVCA 433 cells) or ovarian peritoneal metastasis (HEY cells) we have demonstrated previously in vitro enhanced expression of drug-resistant markers such as ERCC1 or β-tubulin type III in response to short-term cisplatin or paclitaxel treatments [13]. These markers are known to be expressed in tumor samples resistant to platinum or taxane-based therapies [47, 48]. The fact that the enhancement in the expression of these markers coincided with enhanced CSC-like features in ovarian cancer cells strongly suggests an overlap between the chemoresistant features and CSC-like characteristics in ovarian carcinomas.
We observed that cisplatin treatment enhanced the expression of CSC-like markers (EpCAM, CD117, CD44, CD133) in residual ovarian cancer cells. Enhanced expression of these markers in ovarian cancer cells have been shown to be tumorigenic in mouse models [14, 18, 49]. In addition, tumors isolated from ovarian cancer patients at relapse have been shown to have significantly enhanced expression of these markers compared to primary tumors [13, 14]. These findings provide preclinical support for the association of these markers with chemoresistance and recurrence, suggesting CSCs as an attractive target population for novel treatment modalities, whose eradication should potentially lead to permanent tumor regression.
Activation of JAK2/STAT3 has been shown to confer chemo and radioresistance by inducing stem cell-like phenotypes in many cancers including breast [50], prostrate [51], thyroid [52], non-small cell lung [53] and in other cancers. The link between activation of the JAK2/STAT3 pathway and CSCs has also been shown in ovarian cancer, where the stem cell marker CD44 coupled with the embryonic stem cell marker Nanog was linked with the activation of STAT3 in ovarian cancer cells [54]. Such activation of STAT3 in ovarian cancer cells induced the expression of multidrug resistant genes resulting in concomitant chemoresistance. Consistent with these studies, STAT3 pathway activation has been shown to be required for the proliferation and maintenance of glioblastoma stem cells [55], as well as rapidly cycling intestinal stem cells [56]. In addition, LIF and IL-6 mediated STAT3 dependent regulation of the Oct4-Nanog circuitry has been shown to be necessary to maintain the pluripotent inner cell mass, the source of embryonic stem cells [57]. These studies suggest a close relationship between the cytokine-mediated activation of the JAK2/STAT3 pathway and the survival of normal, cancer and embryonic stem cells. In that context, IL-6 secretion has been shown to occur in radiation induced DNA damage [58]. Additionally, elevated levels of IL-6 and transforming growth factor beta (TGFβ) together have been shown to confer resistance to erlotinib [59]. We have recently shown enhanced secretion of IL-6 in paclitaxel surviving ovarian cancer cells [60]. The activation of the JAK2/STAT3 pathway by cisplatin in the current study may be mediated by cytokines such as IL-6 to facilitate resistance to apoptotic pressures in response to cisplatin, thus pushing the residuals cells to survive as a chemoresistant phenotype. As we show in this study, activation of JAK2/STAT3 is crucial for the survival of chemotherapy-resistant CSCs which potentially are the drivers of repopulation and the eventual recurrent disease.
In the present study we demonstrate suppression of enhanced CSC-like characteristics observed in ovarian cancer cells after a single dose of cisplatin treatment in combination with CYT387 in vitro, and the retention of these characteristics in in vivo mouse xenografts. Tumor xenografts generated from cisplatin and CYT387-treated cells had a lower expression of CA125. Elevated level of CA125 is the hallmark of ovarian cancer diagnosis in patients and is frequently observed on relapse of ovarian cancer [4]. CA125 expression has been shown to regulate the growth, tumorigenesis and metastasis of ovarian cancer cells as knock down of CA125 (deleted N-terminal region) completely abrogated the subcutaneous tumor forming ability of SKOV3 ovarian cancer cells in nude mice [61]. Conversely, the same study showed that ectopic expression of CA125 with intact cytoplasmic tail enhanced ovarian tumor growth and metastases in SCID mice. These findings provide evidence that CA125 plays a critical role in ovarian cancer cell growth, tumorigenesis and metastases. The relatively lower abundance of tumorigenic markers in tumors derived from ovarian cancer cells treated with CYT387 and cisplatin compared to cisplatin-treated cells derived tumors suggest a crucial role of the JAK2/STAT3 pathway in maintaining the tumorigenic phenotype in ovarian cancer. The fact that these characteristics induced by chemotherapy and CYT387 can be translated from in vitro to in vivo mouse xenografts suggest that the initial molecular phenotypic changes induced by treatment modalities on surviving cancer cells are retained in recurrent tumors. This may explain sustained activation of JAK2/STAT3 in isolated CR tumor cells compared to tumor cells derived from CN patients.
One limiting aspect of our model like the previously described paclitaxel model [36], was lack of differences in the invasion pattern observed between control untreated, cisplatin-treated, CYT387-treated and cisplatin plus CYT387-treated cells-derived xenografts. This may occur as the cancer cells only received a single short-term dose of cisplatin and/or CYT387 treatments before inoculation into mice. Such short-term treatments even if induced invasiveness (if any) in vitro was not sustained during the six week tumor development in the in vivo microenvironment. However, we observed a more pronounced effect of CYT387 in combination with cisplatin on tumor cell growth in vivo than in vitro. This may potentially result due to the interruption of tumor promoting paracrine effects induced by tumor growth supporting infiltrating and stromal cells in vivo. In future studies, this aspect of the work needs to be investigated further by systemic administration of cisplatin and/or CYT387 in mice intraperitoneally inoculated with ovarian cancer cells. This is likely to show differences in the invasion pattern imposed by cisplatin and/or CYT387 in vivo.
A great number of studies and clinical trials have been designed for the effective management of ovarian cancer patients in the last decade. However, there still remains a need to identify new agents that will specifically target the sustenance of chemoresistant CSC-enriched cancer cells which are represented highly in CR ovarian tumors and are responsible for the relapse of cancer [13, 14]. In this study we have identified activation of STAT3 pathway which is sustained in ascites-derived CR ovarian tumors. The fact that this activation is observed in the absence of stromal or other infiltrating cells may suggest that activation of this pathway may be inherently maintained in tumor cells at recurrence and may be independent of any paracrine effects mediated by the supporting cells. In addition, using cell line models we demonstrate that JAK2/STAT3 pathway is specifically required for the sustenance of CSC-like cells and that it is retained after chemotherapy treatment both in vitro and in vivo. Inhibition of this pathway by CYT387 in conjunction with chemotherapy is a promising strategy for targeting the residual CSC-like ovarian cancer cells which are retained in CR tumors. As CYT387 is already in Phase III clinical trial for myelofibrotic disorder [42] as well as pancreatic cancer (https://clinicaltrials.gov/ct2/show/NCT02101021?term=momelotinib8rank=1), it is possible to rapidly translate the findings described in this study for ovarian cancer treatment.
The authors declare that they have no competing interest.
KA and AL designed the study, performed the experiments, EC also contributed to the experiments, RL helped with the animal experiments, CB provided reagents and was involved with the discussion of the manuscript. EWT and JKF edited the manuscript. NA conceived the idea, designed the study and contributed to the writing of the manuscript.
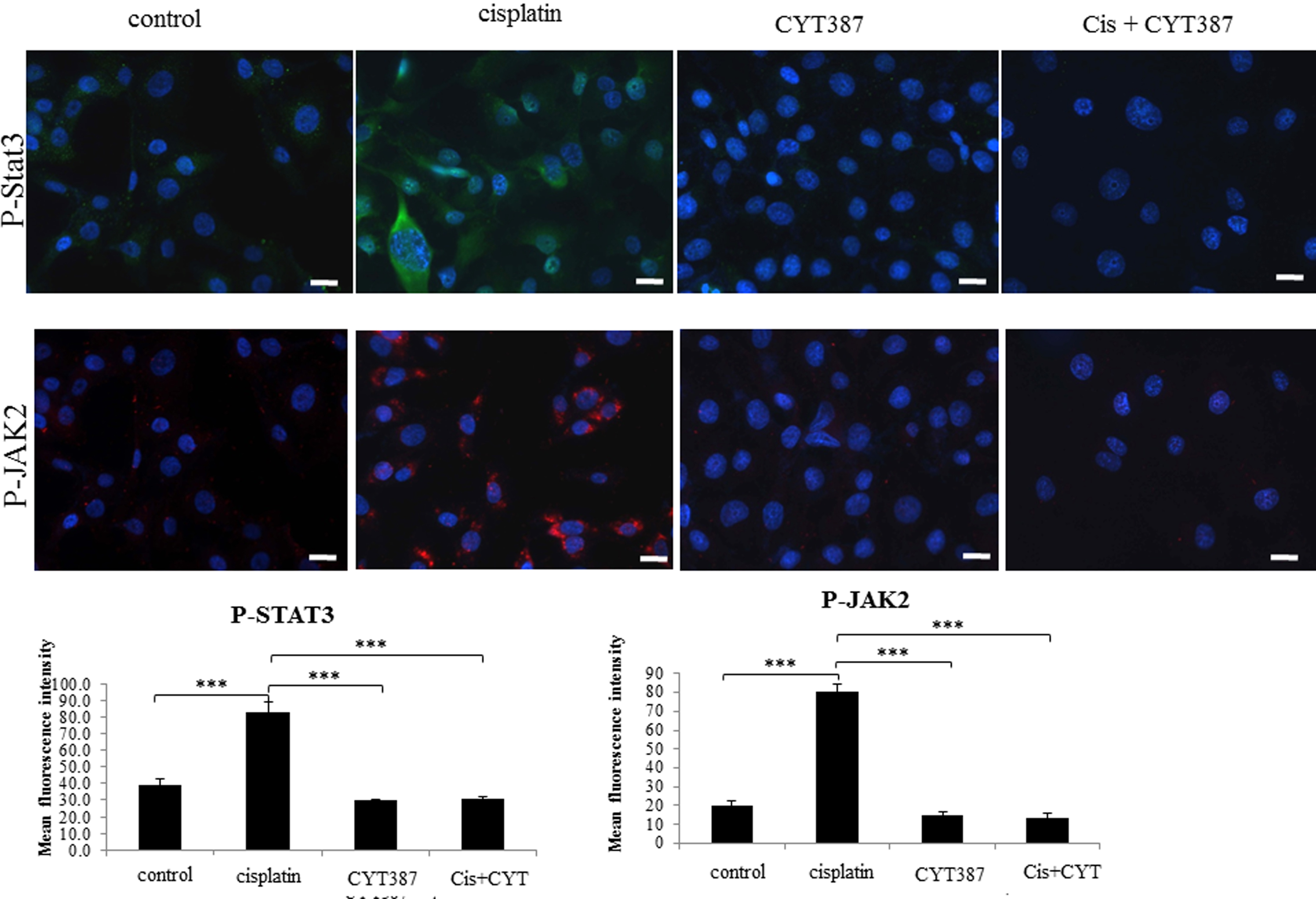 |
Supplementary Figure 1: Expression and localization of P-JAK2 and P-STAT3 in HEY cells in response to cisplatin treatment. The images were evaluated as described in Figure 1. Images are representative of three independent experiments. The mean fluorescence intensity of P-JAK2 and P-STAT3 were quantified using Cell-R software (Olympus Soft Imaging Solutions). Significant intergroup variations are indicated by *** P < 0.001. Magnification 200x; scale bar = 10 μM. |
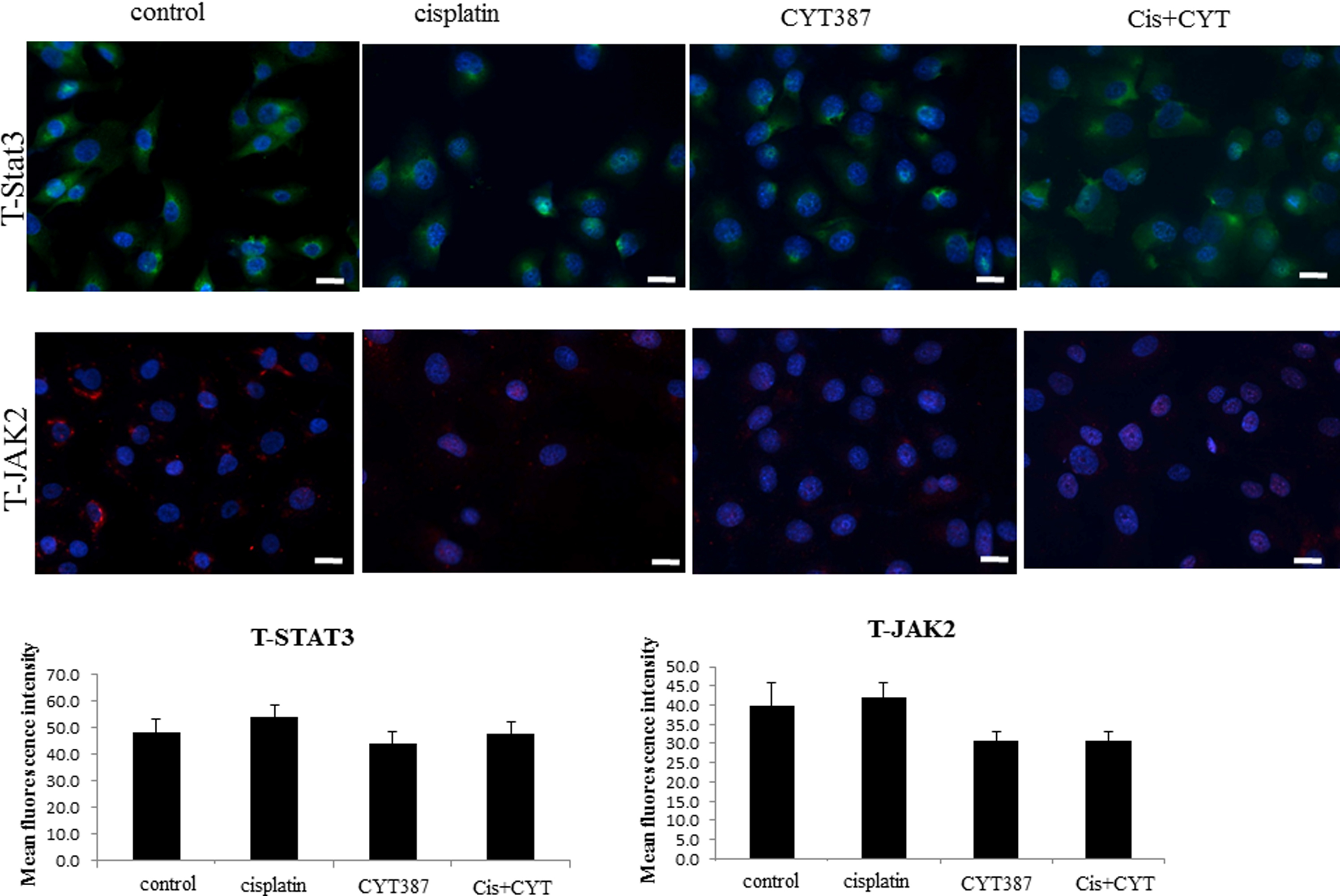 |
Supplementary Figure 2: Expression and localization of T-JAK2 and T-STAT3 in HEY cells in response to cisplatin treatment. The images were evaluated as described in Figure 1. Images are representative of three independent experiments. The mean fluorescence intensity of T-JAK2 and T-STAT3 was quantified using Cell-R software (Olympus Soft Imaging Solutions). Magnification 200x; scale bar = 10 μM. |
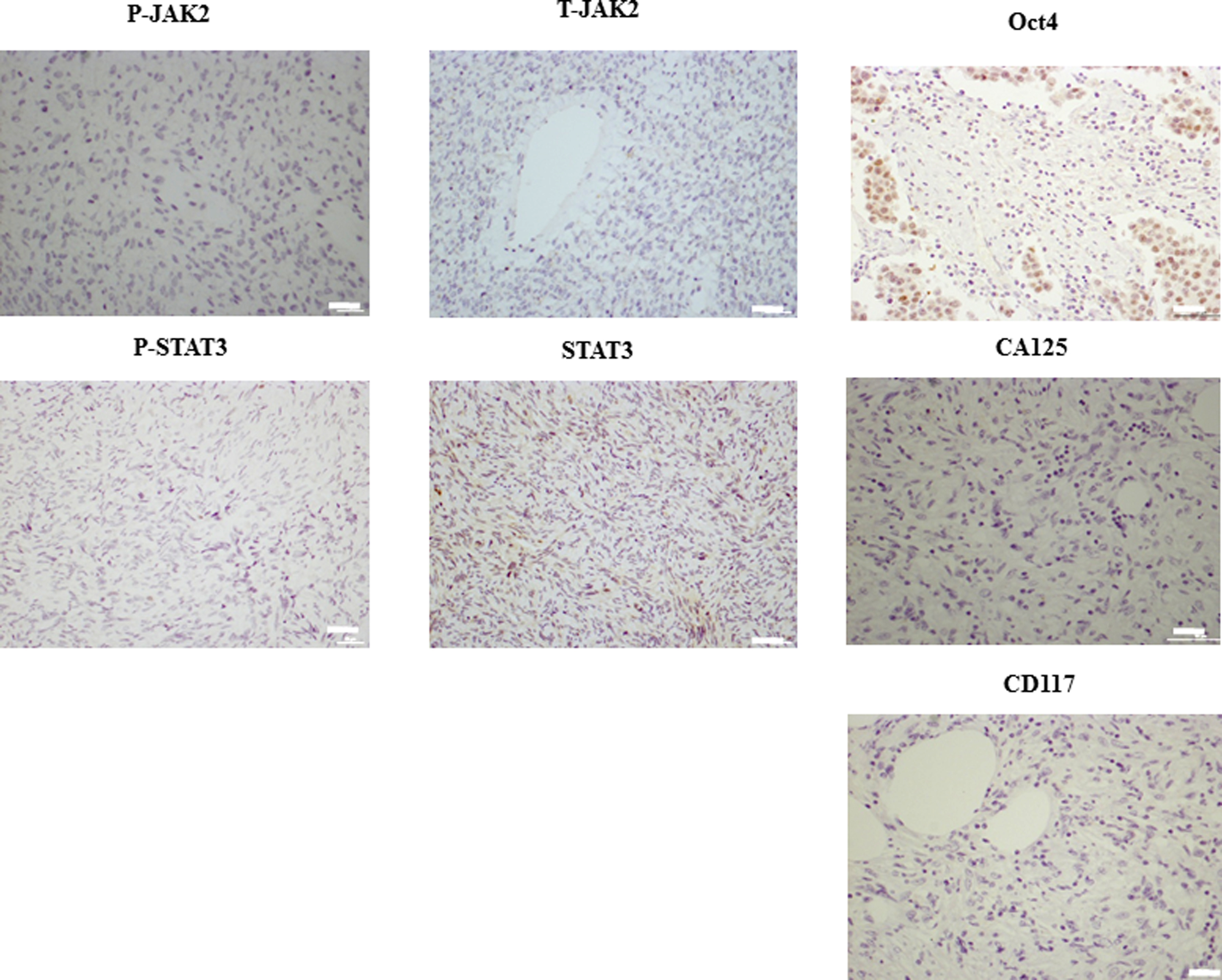 |
Supplementary Figure 3: Negative IgG controls for P-JAK2, T-JAK2, P-STAT3, T-STAT3, Oct4, CD117 and CA125. Tumor sections were stained with IgG control antibodies as described in Methods and Materials. Magnification 200X, scale bar = 10 μm. |
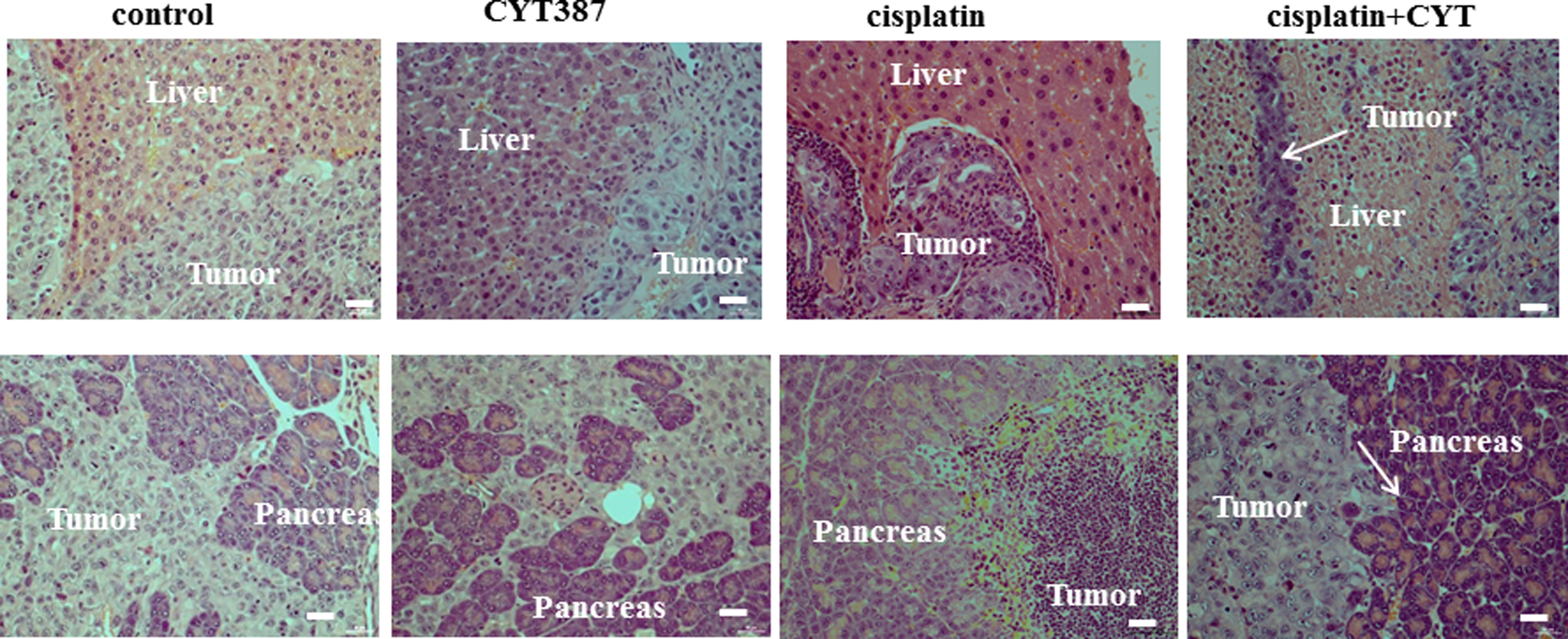 |
Supplementary Figure 4: H and E staining of control and treated HEY cell derived-tumor associated infiltrated organs in mice. 5 × 106 cells were injected ip in each mouse. Histological images of liver and pancreas showing infiltration of control, cisplatin-treated, CYT387 and combination of cisplatin and CYT387-treated (Cis+CYT) HEY cells. Arrows indicate tumor cells invading the respective organs. Magnification 200X, scale bar = 10 μm. |
The authors wish to thank Women's Cancer Foundation, National Health and Medical Research Council of Australia (JKF, RegKey#441101) and the Victorian Government's Operational Infrastructure Support Program and National Breast Cancer Foundation (EWT) for supporting this work. KA and EC are the recipients of Australian Postgraduate Award. AL is a recipient of Royal Women's Hospital Scholarship. RBL is a recipient of the Melbourne Brain Centre Post-Doctoral Research Fellowship from the University of Melbourne. CJB is the recipient of the Dunn Fellowship (Dyson Bequest funding). The authors also wish to acknowledge the help of Dr Simon Nazaretian, Anatomical Pathology, and Royal Women's Hospital for the histological assessment of mouse xenografts.
| [1] | Jemal A, Siegel R, Xu J, Ward E. Cancer statistics 2010. CA Cancer J Clin 2010, 60:277–300. |
| [2] | Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev 2001, 22:255–288. |
| [3] | Lengyel E. Ovarian cancer development and metastasis. Am J Pathol 2010, 177:1053–1064. |
| [4] | Ozols RF. Systemic therapy for ovarian cancer: current status and new treatments. Semin Oncol 2006, 33:S3–11. |
| [5] | Kipps E, Tan DS, Kaye SB. Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat Rev Cancer 2013, 13:273–282. |
| [6] | Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer 2010, 10:803–808. |
| [7] | Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, Traficante N, Fereday S, Hung JA, Chiew YE, Haviv I, Gertig D, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 2008, 14:5198–5208. |
| [8] | Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009, 9:265–273. |
| [9] | Aguilar-Gallardo C, Rutledge EC, Martinez-Arroyo AM, Hidalgo JJ, Domingo S, Simon C. Overcoming Challenges of Ovarian Cancer Stem Cells: Novel Therapeutic Approaches. Stem Cell Rev 2012, 8:994–1010. |
| [10] | Latifi A, Abubaker K, Castrechini N, Ward AC, Liongue C, Dobill F, Kumar J, Thompson EW, Quinn MA, Findlay JK, Ahmed N. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J Cell Biochem 2011, 112:2850–2864. |
| [11] | Abubaker K, Latifi A, Luwor R, Nazaretian S, Zhu H, Quinn MA, Thompson EW, Findlay JK, Ahmed N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol Cancer 2013, 12:24. |
| [12] | Ahmed N, Abubaker K, Findlay J, Quinn M. Cancerous ovarian stem cells: obscure targets for therapy but relevant to chemoresistance. J Cell Biochem 2013, 114:21–34. |
| [13] | Latifi A, Luwor RB, Bilandzic M, Nazaretian S, Stenvers K, Pyman J, Zhu H, Thompson EW, Quinn MA, Findlay JK, Ahmed N. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: molecular phenotype of chemoresistant ovarian tumors. PLoS One 2012, 7:e46858. |
| [14] | Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, Zhang K, Conner M, Landen CN. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res 2012, 18:869–881. |
| [15] | Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, Dombkowski D, Preffer F, Maclaughlin DT, Donahoe PK. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A 2006, 103:11154–11159. |
| [16] | Vathipadiekal V, Saxena D, Mok SC, Hauschka PV, Ozbun L, Birrer MJ. Identification of a potential ovarian cancer stem cell gene expression profile from advanced stage papillary serous ovarian cancer. PLoS One 2012, 7:e29079. |
| [17] | Zeimet AG, Reimer D, Sopper S, Boesch M, Martowicz A, Roessler J, Wiedemair AM, Rumpold H, Untergasser G, Concin N, Hofstetter G, Muller-Holzner E, Fiegl H, Marth C, Wolf D, Pesta M, et al. Ovarian cancer stem cells. Neoplasma 2012, 59:747–755. |
| [18] | Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68:4311–4320. |
| [19] | Alvero AB, Chen R, Fu HH, Montagna M, Schwartz PE, Rutherford T, Silasi DA, Steffensen KD, Waldstrom M, Visintin I, Mor G. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8:158–166. |
| [20] | Alvero AB, Fu HH, Holmberg J, Visintin I, Mor L, Marquina CC, Oidtman J, Silasi DA, Mor G. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells 2009, 27:2405–2413. |
| [21] | Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci 2000, 25:496–502. |
| [22] | Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron-Smith D, Findlay J, Ackland L, Ahmed N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer 2009, 100:134–144. |
| [23] | Huang WL, Yeh HH, Lin CC, Lai WW, Chang JY, Chang WT, Su WC. Signal transducer and activator of transcription 3 activation up-regulates interleukin-6 autocrine production: a biochemical and genetic study of established cancer cell lines and clinical isolated human cancer cells. Mol Cancer 2010, 9:309. |
| [24] | Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21:2000–2008. |
| [25] | Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol Sci 2005, 26:113–116. |
| [26] | Liu JJ, Lu J, McKeage MJ. Membrane transporters as determinants of the pharmacology of platinum anticancer drugs. Curr Cancer Drug Targets 2012, 12:962–986. |
| [27] | Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev 2012, 64:706–721. |
| [28] | Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31:1869–1883. |
| [29] | Jazaeri AA, Awtrey CS, Chandramouli GV, Chuang YE, Khan J, Sotiriou C, Aprelikova O, Yee CJ, Zorn KK, Birrer MJ, Barrett JC, Boyd J. Gene expression profiles associated with response to chemotherapy in epithelial ovarian cancers. Clin Cancer Res 2005, 11:6300–6310. |
| [30] | Johnatty SE, Beesley J, Paul J, Fereday S, Spurdle AB, Webb PM, Byth K, Marsh S, McLeod H, Harnett PR, Brown R, DeFazio A, Chenevix-Trench G. ABCB1 (MDR 1) polymorphisms and progression-free survival among women with ovarian cancer following paclitaxel/carboplatin chemotherapy. Clin Cancer Res 2008, 14:5594–5601. |
| [31] | Peters D, Freund J, Ochs RL. Genome-wide transcriptional analysis of carboplatin response in chemosensitive and chemoresistant ovarian cancer cells. Mol Cancer Ther 2005, 4:1605–1616. |
| [32] | Jayachandran G, Ueda K, Wang B, Roth JA, Ji L. NPRL2 sensitizes human non-small cell lung cancer (NSCLC) cells to cisplatin treatment by regulating key components in the DNA repair pathway. PLoS One 2010, 5:e11994. |
| [33] | Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest 2007, 117:1370–1380. |
| [34] | Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Saito M, Kawagoe J, Takahashi K, Yada-Hashimoto N, Sakata M, Motoyama T, Kurachi H, Tasaka K, Murata Y. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem 2004, 279:23477–23485. |
| [35] | Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett 2007, 251:1–16. |
| [36] | Abubaker K, Luwor RB, Zhu H, McNally O, Quinn MA, Burns CJ, Thompson EW, Findlay JK, Ahmed N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014, 14:317. |
| [37] | Buick RN, Pullano R, Trent JM. Comparative properties of five human ovarian adenocarcinoma cell lines. Cancer Res 1985, 45:3668–3676. |
| [38] | Abubaker K, Luwor RB, Escalona R, McNally O, Quinn MA, Thompson EW, Findlay JK, Ahmed N. Targeted Disruption of the JAK2/STAT3 Pathway in Combination with Systemic Administration of Paclitaxel Inhibits the Priming of Ovarian Cancer Stem Cells Leading to a Reduced Tumor Burden. Front Oncol 2014, 4:75. |
| [39] | Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther 2004, 10:1032–1042. |
| [40] | Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol 2013, 3:256. |
| [41] | Duan Z, Foster R, Bell DA, Mahoney J, Wolak K, Vaidya A, Hampel C, Lee H, Seiden MV. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin Cancer Res 2006, 12:5055–5063. |
| [42] | Pardanani A, Laborde RR, Lasho TL, Finke C, Begna K, Al-Kali A, Hogan WJ, Litzow MR, Leontovich A, Kowalski M, Tefferi A. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia 2013, 27:1322–1327. |
| [43] | Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010, 115:5232–5240. |
| [44] | Monaghan KA, Khong T, Burns CJ, Spencer A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia 2011, 25:1891–1899. |
| [45] | Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC, Reibel JB, Tamayo P, Godfrey JT, Qian ZR, Page AN, Maciag K, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov 2014, 4:452–465. |
| [46] | Ahmed N, Abubaker K, Findlay J, Quinn M. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr Cancer Drug Targets 2010, 10:268–278. |
| [47] | Muallem MZ, Braicu I, Nassir M, Richter R, Sehouli J, Arsenic R. ERCC1 expression as a predictor of resistance to platinum-based chemotherapy in primary ovarian cancer. Anticancer Res 2014, 34:393–399. |
| [48] | Kavallaris M, Kuo DY, Burkhart CA, REgl DL, Norris MD, Haber M, Horwitz SB. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest 1997, 100:1282–1293. |
| [49] | Meirelles K, Benedict LA, Dombkowski D, Pepin D, Preffer FI, Teixeira J, Tanwar PS, Young RH, MacLaughlin DT, Donahoe PK, Wei X. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc Natl Acad Sci U S A 2012, 109:2358–2363. |
| [50] | Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, Bessarabova MO, Huh SJ, Silver SJ, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest 2011, 121:2723–2735. |
| [51] | Hellsten R, Johansson M, Dahlman A, Sterner O, Bjartell A. Galiellalactone inhibits stem cell-like ALDH-positive prostate cancer cells. PLoS One 2011, 6:e22118. |
| [52] | Tseng LM, Huang PI, Chen YR, Chen YC, Chou YC, Chen YW, Chang YL, Hsu HS, Lan YT, Chen KH, Chi CW, Chiou SH, Yang DM, Lee CH. Targeting signal transducer and activator of transcription 3 pathway by cucurbitacin I diminishes self-renewing and radiochemoresistant abilities in thyroid cancer-derived CD133+ cells. J Pharmacol Exp Ther 2012, 341:410–423. |
| [53] | Hsu HS, Huang PI, Chang YL, Tzao C, Chen YW, Shih HC, Hung SC, Chen YC, Tseng LM, Chiou SH. Cucurbitacin I inhibits tumorigenic ability and enhances radiochemosensitivity in nonsmall cell lung cancer-derived CD133-positive cells. Cancer 2011, 117:2970–2985. |
| [54] | Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem 2008, 283:17635–17651. |
| [55] | Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27:2383–2392. |
| [56] | Matthews JR, Sansom OJ, Clarke AR. Absolute requirement for STAT3 function in small-intestine crypt stem cell survival. Cell Death Differ 2011, 18:1934–1943. |
| [57] | Do DV, Ueda J, Messerschmidt DM, Lorthongpanich C, Zhou Y, Feng B, Guo G, Lin PJ, Hossain MZ, Zhang W, Moh A, Wu Q, Robson P, Ng HH, Poellinger L, Knowles BB, et al. A genetic and developmental pathway from STAT3 to the OCT4-NANOG circuit is essential for maintenance of ICM lineages in vivo. Genes Dev 2013, 27:1378–1390. |
| [58] | Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 2009, 11:973–979. |
| [59] | Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, Kenner L, Sordella R. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A 2010, 107:15535–15540. |
| [60] | Ahmed N, Abubaker K, Findlay JK. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol Aspects Med 2014, 39:110–125. |
| [61] | Theriault C, Pinard M, Comamala M, Migneault M, Beaudin J, Matte I, Boivin M, Piche A, Rancourt C. MUC16 (CA125) regulates epithelial ovarian cancer cell growth, tumorigenesis and metastasis. Gynecol Oncol 2011, 121:434–443. |