| Journal of Cancer Stem Cell Research (2014), 2:e1007 © 2014 Creative Commons. All rights reserved ISSN 2329-5872 DOI: 10.14343/JCSCR.2014.2e1007 http://cancerstemcellsresearch.com |
|
| Journal of Cancer Stem Cell Research (2014), 2:e1007 © 2014 Creative Commons. All rights reserved ISSN 2329-5872 DOI: 10.14343/JCSCR.2014.2e1007 http://cancerstemcellsresearch.com |
|
| Review Article | Open Access |
| Glioblastoma stem-like cells: approaches for isolation and characterization | |
| Erika S. Molina1, Micheli M. Pillat1, Vivaldo Moura-Neto2, Tamara T. Lah3, and Henning Ulrich1,* | |
| 1Departmento de Bioquímica, Instituto de Química, Universidade de São Paulo, São Paulo, Brazil, 2Instituto Estadual do Cérebro Paulo Niemeyer, Rio de Janeiro, Brazi, 3Department of Genetic Toxicology and Cancer Biology, National Institute of Biology, Ljubljana, Slovenia and Faculty of Chemistry and Chemical Engineering, University of Ljubljana, Slovenia | |
| *Corresponding author: Tel.: +55 11 30918512 E-mail address: henning@iq.usp.br Departamento de Bioquímica, Instituto de Química, Universidade de São Paulo Av. Prof. Lineu Prestes 748, São Paulo, 05508-000 SP, Brazil Tel.: +55 11 30919181 Received: September 8, 2014; Revised: October 21, 2014; Accepted: October 22, 2014 | |
Abstract: Glioblastoma (GBM) stem-like cells (GSCs) represent the most undifferentiated state of malignant cells with distinct biological characteristics. The fraction of these cells within a glial brain tumor, ranging from 2–30%, is correlating with the increasing WHO stage and poor prognosis of patients' survival. GSCs represent the least vulnerable, thus most preferential target cell population to be exposed to various therapeutic modalities, although the underlying mechanisms of this resistance are not yet fully understood. For the development of GSC-targeting therapies, further in depth studies are needed using enriched and stable GSC cell populations. Here, we discuss the current approaches of GSC isolation and validation based on expression of stemness and oncogenic markers as well as on functional assays. The enrichment of GSC phenotypes in established cell lines and/or primary tumor cultures, achieved by different strategies, is reviewed, providing a comprehensive comparison of selected studies and contemplating the characterization of the plethora of variants of reported GBM population exhibiting the GSC phenotype.
Keywords: Brain tumors, Glioblastomamultiforme,Glioblastoma stem-like cells,Cancer biomarkers, Tumormicroenvironment.
Aldehyde dehydrogenase (ALDH)
Brain tumor initiating cells (BTIC)
Cancer stem cell (CSC)
Epidermal growth factor (EGF)
Epithelial to mesenchymal transition (EMT)
Fibroblast growth factor (FGF)
Glioblastoma multiforme (GBM)
Glioblastoma stem-like cell (GSC)
Iso-dehydrogenase 1 (IDH1)
Neural progenitor cell (NPC)
Neural stem cell (NSC)
O-6-methylguanine-DNA-methyltransferase (MGMT)
Temozolomide (TMZ)
The most common primary brain tumors are derived from genetic anomalies in glial development in the brain, comprising of astrocytoma and oligodendroglioma [1, 2]. These are graded on a scale with increasing malignancy as lower grade astrocytoma (WHO I and II), anaplastic astrocytoma (WHO III) and glioblastoma multiforme (GBM; WHO IV), also termed glioblastoma (GBM). GBM is the most aggressive of these tumors and ranks among the most desperate of all human cancers. Survival of GBM patients, although individually variable, has improved from an average of 10 to only 14 months after diagnosis in the last 5 years, in spite of significant improvements in the standard care with more targeted therapy (reviewed in [3–5]). In addition to several clinical and histopathological parameters, as well as specific therapeutic approaches, patients' responses to therapy are heterogeneous. This is related to insufficient data for prognostic and survival stratification. More information about molecular fingerprints of tumors as well as to better understanding of cellular origin of the tumor-initiating cells will help to develop individualized therapies for patients.
The classification of malignant gliomas is changing rapidly, and novel schemes are being developed based on their genetic landscapes [6] from next generation sequencing [7, 8], followed by epigenetics, proteomics and oncogenic miRNA data [9]. These are superimposed on the histological origin of brain tumor-initiating cells (BTIC) and their precursors evolving into glioma stem-like cells, also termed glioma stem cells (GSC). Thus, in about 90% of cases, GBMs arise de novo (primary GBM) without any evidence of less malignant precursor lesions. Secondary GBMs progress from low-grade diffuse astrocytoma or anaplastic astrocytoma [2], occurring in younger patients and having a lesser degree of necrosis and significantly better prognosis. Although primary and secondary GBMs are nearly indistinguishable in their histology, they differ in their genetic and epigenetic profiles providing the state-of-the-art GMB classification. Decisive genetic markers for secondary GBMs are iso-dehydrogenase 1 (IDH1) mutations, which are absent in primary GBMs and associated with a hypermethylation (particularly of histones) phenotype. IDH1 mutations are the earliest detectable genetic alterations in precursors of low-grade diffuse astrocytomas and in oligodendrogliomas. The former acquire further TP53 and ATRX mutations, whereas the latter lose the 1p/19q arm and are mutated in CIC and FUBP1 genes. In contrast, primary GBM glial progenitor cells overexpress epidermal growth factor (EGF) receptors and exhibit TP53 and PTEN mutations as wells as LOH 10p and LOH 10q deviations. Therefore, recent classification of glioma is based on the tumor metabolome and epigenetic changes. As an example, methylation of the O-6-methylguanine-DNA-methyltransferase (MGMT) promoter silences expression of this gene by 40% in the primary tumor, and more than 70% in the secondary GBM [7]. As this enzyme acts by de-methylating alkyl groups, epigenetic silencing of its expression increases GBM sensitivity to treatment by DNA-alkylating agents, such as commonly used temozolomide (TMZ) [10].
GBMs are histologically similar and featured by atypical mitotic nuclei often associated with necrosis and/or angiogenesis. The standard treatment against GBM includes surgery, radiotherapy and, in the last 5 years, chemotherapy with TMZ [11]. However, since a population of the invasive cells is dispersed through surrounding normal tissue, and GBMs reveal resistance against radio- and chemotherapy, the effectiveness of the multimodal treatment is limited, resulting in GBM reoccurrence.
The observed diversity of molecular fingerprinting might originate from the genetically different glioma initiating cells [3] also called cancer stem-like cells, here termed glioblastoma stem cells (GSCs). Similar cells have been found in many, although not in all human tumors. These “roots of cancer” operate in a hierarchical fashion such as conceptualized for normal stem cells [12]. The authors of this work reviewed the evolution of the cancer stem cell (CSC) model, which is going back to the 19th century to Virchow's embryonic-rest hypothesis, where cancer is a consequence of the activation of ‘dormant’ stem-like cells in adult tissues. The basic understanding of the current cancer stem cell theory is that heterogeneity within the tumors is not a mere consequence of random mutations and selection, leading to well recognized clonal evolution of cancer [13], but results from an intrinsic hierarchy of tumor cells [14–16]. CSCs have unlimited capability to divide symmetrically and asymmetrically. Asymmetric cell division occurs for originating progenitor cells together with a reduction in the differentiation potential. The theory of a hierarchical model of tumorigenesis has been widely accepted, thus stating that only a small fraction of tumor cells - the CSCs, are capable of initiating tumor growth or renewing the tumor in the same or other organ after incomplete surgical removal [17, 18]. When injected orthotopically, CSCs are capable of forming tumors in animal. Likewise, CSCs can directly or indirectly contribute to generation of metastasis [19]. However, the existence and identification of these cells remains an open question, as all the existing in vitro and in vivo using animal models in fact represent artificial environments. Consequently it cannot be ruled that the so named CSCs are just cells which have adapted to altered growth conditions.
The above-described CSC model has a “static” hierarchical structure, largely based on the notion that normal stem cells undergo oncogenic transformation. However, in the past few years a number of publications have challenged this concept by demonstrating that in the tumor environment progenitors, differentiated cells and even transformed tumor cells may acquire the ability of self-renewal through their de-differentiation, e.g. a reversal of differentiation [20, 21], reviewed in [22], or by other mechanisms, discussed in the following [23]. Moreover, the finding that epithelial to mesenchymal transition (EMT), which is a reversible process and can transiently induce stem-like cells phenotypes, was crucial to expand static the CSC model to the fluid CSC model [12], also termed the CSC plasticity model. This model proposes CSC plasticity, meaning that by dedifferentiation of a variety of cell types within growing tumor mass even various populations of CSCs may evolve by acquiring additional mutations or epigenetic modifications. Several authors propose that various CSC subclones can exist as the result of evolutionary pressure of therapies, giving rise to more aggressive secondary CSCs [24]. Taken together, the clonal evolution and CSC hierarchical theory merge into a so called fluid plasticity or convergence CSC model. This results in several subclones of CSC within the same tumors, depending on epigenetic and mutational events in specific cells with self-renewal capabilities. These secondary aggressive CSCs may become dominant and drive tumor formation or simply co-exists with other phenotypes of CSCs. Such concepts represent challenges in order to develop novel therapeutic approaches that would more efficiently target the cancerous clonal population with stem cell-like characteristics.
Recent research has revealed that GBM stem-like cells play important roles in GBM pathogenesis. The first evidence that GBM display a cellular hierarchy with self-renewing and tumor-initiating cell types located at the apex was provided by Singh et al [25, 26] and Galli et al [27] followed by further relevant studies on this topic (reviewed in [28–30]. The origin of GBM stem cell has been widely discussed and is now based on the fact that genome analyses revealed the transcriptional fingerprints of neural stem cells (NSC), their progenitors, as well as of mesenchymal cells [31, 32]. Tumor initiating cell based classification comprises four subtypes, such as classical, mesenchymal, neural and proneural transcription profiles, as summarized by Van et al. [3]. These GBM stem-like cells (GSCs) self-renew, their hijacking molecular mechanisms and expressing markers of NSCs [7, 33]. Over the past decade, a wide range of studies have shown that several signaling proteins involved in neural development also play important roles in GBM pathogenesis, as highlighted and discussed in the context of developing treatments for GBM [34]. GSCs express the intermediate filament protein nestin, the transcription factors Sox2 and Oct 3/4, the RNA-binding protein musahi, transcriptional repressor Bmi1 of the polycomb of proteins, proteins of the Notch signaling pathway receptor, aldehyde dehydrogenase 1 (ALDH1) and among a few others also the extensively studied transmembrane protein, prominin-1 (CD133) (reviewed in [35, 36]). It has been suggested that the resistance of GBMs towards radiation and chemotherapy is attributed to the GSC-like population [28, 37, 38]. Hence, GSCs are proposed to persist as a distinct population involved in tumor relapse and propagation and significantly impact the poor prognostics of the GBM patients. Accordingly, GSCs have been pursued as a promising target for the development of novel specific diagnosis/prognosis therapies against GBM [39].
Although GSCs are increasingly studied for molecular mechanisms, immunophenotyping or targeting purposes, the GSC markers, functional assays and the culture conditions remain controversial challenging further investigations [40]. Different methods of GSC enrichment have been proposed, either from GBM cell lines and/or primary cultures [41], resulting in GSCs with rather different phenotypes. Thus, the enrichment of GSCs by itself may represent an artifact and contribute to misidentification of these cells and to the discrepancies in their identification and characterization.
Flow cytometry and fluorescence-activated cell sorting (FACS) provide the opportunity to analyze various parameters in a single live cell as well as separating heterogeneous cell populations based on these characteristics. Besides differential epitope expression, morphological properties, such as cell size and granularity measured as forward and side scatter parameters are assessed. Similar to the identification of embryonic and adult stem cells, measurements of specific clusters of cell surface markers and enzymes responsible for metabolic activities are the methods of choice in flow cytometric identification of tumor stem cells, as illustrated in Figure 1 and detailed below. However, the reliability of these tumor cell markers remains questionable. For instance, tumor cells in general are known for genetic instability, and consequently proteome profiles of GSC biomarkers may be difficult to analyze. Furthermore, epigenetic changes, induced by tumor microenvironment, in particular when created in the so called stem cell niches, induce expression of multipotency genes in GSCs [20, 23, 42]. It is worthwhile mentioning that phenotypes may change under in vitro culture conditions (reviewed in [43]).
 |
Figure 1. GSC represent a promising target for treating GBM. Classical GSC enrichment consists of serial transplantation in vivo and/or culture with defined medium supplemented with EGF and FGF-2, which might be improved by mimicking the micro-environment niche of GSC through hypoxia and/or co-cultivation with endothelial cells. GSC isolation by FACS has been based on differential expression of CD133 followed by ALDH (Aldefluor) as more contemplating functional assessment of GSC phenotype. Additionally, aptamers have turned into promising tools for targeting biomarkers allowing identification of specific binders against GSC populations. |
As discussed above, GSCs are highly plastic. Consequently similar clones with different biomarker fingerprints without similar characteristics may exist even in the same tumor tissue. Nevertheless, the strategies for CSC identification, isolation and targeting discussed below, have led to our current knowledge and novel strategies of glioblastoma. To date, multi-parameter flow cytometry has been developed for detection of a combination of characteristics, which define a rare population, such as cancer stem cells or circulating tumor cells in the blood [44, 45]. Such rare populations can be identified in a single experiment, following staining for side populations (Hoechst dye exclusion by ABC transporters), ALDH1 in the presence of a substrate, which becomes fluorescent following enzymatic cleavage, and CD markers, i.e. CD133, and exclusion of dead cells (i.e. stained by 7-aminoactinomycin-D, 7-AAD), such as proposed by Greve and co-workers [43]. Optimization of fluorescence probes as well as more knowledge on selection of molecular targets by screening for expression profiles will improve the identification and separation of live cells. Some of the pluripotency-coding genes, supposedly also expressed by tumor stem cells, are intracellular antigens, which cannot be targeted in live cells by flow cytometry. Therefore, molecular beacons were developed for flow cytometry detection of gene expression of these genes on the mRNA level [46]. Applications of in vivo flow cytometry and advanced imaging together with an automated method for identifying and tracking rare cell events are available for detection of circulating tumor cells in the blood [47]. Galanzha and Zharov developed a highly sensitive method, by which the entire blood volume (5 L) of a patient can be analyzed in vivo by photoacoustic flow cytometry [48]. Further perspectives here are switching from the identification of entire cells to the detection of circulating cancer stem cells and associated microvesicles.
GSCs can be also identified within side population (SP) assays using flow cytometry. This SP cell population is obtained by the efflux of Hoechst fluorescent dyes revealing stem cell-like characteristics. This dye-efflux, presumably due to the exclusion of the dye triggered by an ATP binding cassette (ABC) transporter, was also identified in cells from several tumors exhibiting stem-like genes, tumorigenicity and multi-drug resistance [49, 50]. In the C6 glioma cell line, SP cells are described as enriched in stem-like cells [51]. To date, the SP is employed as gating criteria for GSC enrichment by flow cytometry with following demonstration of self-renewal, multi-lineage differentiation and tumorigenicity [52] or decreased migration [53]. However, as a drawback to GSC enrichment and purification based on Hoechst exclusion, recent evidences indicate that, in fact, the GBM SP cells do not contribute to self-renewal or tumor initiation and thus is not enriched in GSC and not tumorigenic [54] but consists of brain endothelial cells [55]. In fact, recent evidence shows that the three present ABC transporters ABCA1, MRP4 and MRP5 are differentiation instead of stemness markers [56]. Taken together, SP flow cytometry is not recommended as GSC enrichment methodology.
The surface marker prominin-1 CD133 is widely used as phenotypic markers for enrichment of CSCs, including GSCs, although it is also expressed by hematopoietic, endothelial and neural progenitors cells [57–58] showing that in the GBM SC population, membrane bouding expression of CD133 was restricted to tumor cells bearing the extracellular CD133 epitope AC133 [59]. Furthermore, CD133 may also be expressed by differentiated tumor cells with changed conformation as result of differential glycosylation, thus masking the epitope recognized by the anti-AC1333 antibody [59, 60]. CD133 expression is discussed controversially, regarding its use as criterion for GSC enrichment, since GBM tumorigenesis is not only driven by CD133-positive cells. Indeed, some CD133-negative cells comprise the most undifferentiated and aggressive cells placed in the apex of the hierarchy [61]. In view of that, the polycomb protein EZH2 has been indicated to be a more specific marker for CSCs than CD133 [62]. The enrichment of CSCs or GSCs based on EZH2 expression remains to be proven.
Aldehyde dehydrogenases (ALDH) are a family of cytosolic NAD(P)+-dependent enzymes that catalyze the oxidation of a wide spectrum of endogenous and exogenous aliphatic and aromatic aldehyde substrates to their corresponding carboxylic acids [63]. High expression of isoenzyme ALDH-1 was for long correlated with resistance to highly reactive alkylating agent based therapies, such as derivatives of cyclophosphamide. The enzyme detoxifies alkylating agents by converting them into less reactive molecules and favoring survival of leukemia tumor [64] and GBM cell populations [65, 66]. ALDH-1 has been proposed to also mediate the resistance of GBM against the current standard chemotherapy compound TMZ [67]. Consequently, ALDH inhibition enhances cytotoxicity of alkylating agents for GSCs [68]. ALDH-2 and ALDH-3 prevent cytotoxicity such as ALDH-1 [65]. However, only ALDH-1A1 is involved in the metabolic oxidation of retinal, also known as retinaldehyde or vitamin A aldehyde, to the active metabolite retinoic acid (RA) [69]. Retinal promotes differentiation of GSCs, although it is not sufficient to antagonize differentiation inhibition by growth factors such as EGF and fibroblast growth factor (FGF)-2 [70]. ALDH-1 has thereby been proposed as a CSC and GSC marker [71–73]. Interesting, hypoxia induces or up regulates ALDH-1 expression in established and primary GBM cell lines [74]. Controversially, ALDH-1A1expression was also considered a marker of astrocytic differentiation in brain tumors correlating with longer survival of GBM patients [75].
Membrane-permeable fluorescent substrates for ALDH, such as i.e. dansyl aminoacetaldehyde (DAAA) allow identification and isolation of ALDH positive cells by flow cytometry [76]. However, the DAAA-based method does not only require further fractionation steps to yield highly purified stem cells and dye excitation with UV light could be mutagenic to the purified cells. A better alternative is the stable fluorochrome Bopidy aminoacetaldehyde diethylacetal (BAAA-DA), which when converted into the fluorescent ALDH substrate (BAAA) is excited by visible light, yielding brighter emission than other fluorochromes, which could be detected using green fluorescence (FL-1) channel of a standard flow cytometer [77]. Hence, Aldefluor assays can be used in combination with other fluorescent labels such as R-phycoerythrin (PE), PE-tandem conjugates, PerCP, Cyanine-5 (Cy-5) and allophycocyanin (APC), which are detected in the FL-2, FL-3 or FL-4 channels. Aldefluor assays employ the ALDH inhibitor diethylamino-benzaldehyde (DEAB) to set up the fluorescent background of negative control populations [78]. In addition, due to the highly expression of the multiple drug resistance (MDR) pump by stem-like cells [79]. Aldefluor assays also contain a MDR inhibitor to avoid the efflux of the fluorescent product better resolving ALDH positive from negative discrete cell populations [78, 80]. Remarkably, all refinement provided by Aldefluor assays are improving ALDH-activity studies by fluorescence-activated cell sorting (FACS), supporting them as a tool marker for isolation of NSCs [81] and GSCs [82, 83] as well as for other CSCs [84, 85]. ALDH-1A2 and ALDH-2 activities are also measured by the Aldefluor assay [86].
Despite the ongoing controversial discussion on the diagnostic value of tumor stem cell markers and whether stem cells are a stable component of the tumor cell population and not merely a transient phenotype within a certain tissue niche, attempts have been made to diagnostically and therapeutically target the above-discussed marker epitopes by high-affinity and specificity ligands. In general, cell surface and transmembrane proteins expressed by various types of tumor stem cells, including CD44, CD47, CD123, EpCAM (CD326), CD133 and IGF receptor I, as well as antibodies interfering with notch-receptor signaling have been used for identification of monoclonal antibodies of possible therapeutic value (reviewed by Naujokat, 2014).
Aptamers, evolved from combinatorial DNA or RNA oligonucleotide libraries by an in vitro selection process called SELEX, systematic evolution of ligands by exponential enrichment) against a target epitope compete with antibodies for many applications. Their synthetic nature, not involving animals in any process, as well as the easiness of chemical modifications for increasing stability in biological solutions including plasma have turned them into promising agents for diagnosis and therapy [88, 89, 90]. There are two alternatives in using aptamers for identification and isolation of GSCs: (1) Selection of aptamers against their putative biomarkers using purified or recombinant expressed proteins. (2) Developing aptamers against GSC-like cells by Cell SELEX. This recently developed technique uses intact live cells expressing the target epitope(s) and exposes them to the combinatorial library for aptamer selection (reviewed in [91]). The differences between the epitopes expression on the cell surface of a tumor-stem like cells, vs. an untransformed or non-stem cell of the same histologic origin can be exploited for obtaining tumor stem cell-specific aptamers.
Aptamers were created with success as specific binders of GBM populations. First, aptamers following reiterative cycles of Cell-SELEX were identified binding to human GBM cell lines. Kang and co-workers [92] first used the procedure to isolate GBM cells, obtaining a set of aptamers not interacting with non-neoplastic astrocytes and other tumor types. Another approach to identify GBM cells was to raise aptamers against the EGF variant III (EGFRvIII), one of the most common mutants in GBM [93]. These aptamers were used as diagnostic tools, along with highly specific radionuclide molecular imaging of GBM in vivo [94]. However, they were developed for targeting a heterogeneous tumor population and not stem cells within this cell mixture.
The development of nuclease-resistant 2′-fluoropyrimidine-modified RNA aptamers, which recognize CD133 and AC133 epitopes with high specificity [95] is a significant progress towards targeting CSCs. DY647-fluorescence labelled anti-CD133 aptamers were verified regarding their diagnostic potential in imaging and flow cytometry. Furthermore, these aptamers were effective in penetrating tumorspheres and internalized by tumor cells, opening a possibility for loading of the aptamer with a cytotoxic compound or si-RNA for down-regulation of oncogenes relevant for cell survival. Along the same line, Kim et al. [96] enriched CD133-positive cells by immunobead separation from mice GSC xenografts, and validated these for their tumor-initiating capacity in another animal model. Then, the authors used CD133-negative cells as well as non-neoplastic neural progenitor cells as negative control for a subtraction process for removal of all common aptamer binders to both cell types. As already shown for the aptamers selected by Shigdar et al. [95], the aptamers bound to proliferating and tumor-initiating cells with dissociation constants in the subnanomolar range. The authors concluded that these aptamers may gain therapeutic importance, if conjugated to tacytotoxin. Indeed, paclitaxel-loaded nanoparticles, reducing GBM proliferation [97], have been shown to cross the blood brain barrier [98]. In addition, aptamer penetration of brain tumors can be facilitated by liposomes, such as those used as vehicles for doxorubicin-loaded aptamers into brain tumors in animal models [99, 100].
Since the first experimental studies supporting in vitro self-renewal and capacity of GSCs to differentiate into neuronal and glial cells, GSC enrichment has been essentially performed in vitro using defined medium. Originally, GSCs were identified by their capacity to form three dimension cellular clusters, termed tumorspheres, similar to neurosphere cultures of NSCs and NPCs [101, 102]. Spheres culture is considered to keep the undifferentiated state of the cells by minimizing stimulation from the environment, such as adhesion to substrates, but also avoiding the access of the cells in the core of the sphere to differentiation factors [103]. Classically, GSCs arive in adherent cultures with defined medium following 20 to 40 days after plating [25, 27, 104, 105]. Based on this, tumorspheres formation has been obtained faster through anchorage-independent culture, such as in dishes previously treated with poly-Hema (poly2-hydroxyethyl methacrylate) [105, 106], gelatin [83] and agar [107]. In fact, tumorigenicity correlates with suspension growth [108]. In addition, three-dimensional culturing models are also available, including gelatin foam cultures, for the study of GSCs [109]. However, special care should be taken regarding suspension cultures [110], since poly-Hema derived spheres might not be truly three dimensional clone derived, but rather reflect cell aggregates. In fact, the formation of spheres as predicative property of CSC enrichment has been questioned, as no correlation was found with the expression of GSC markers [111]. Indeed, the technique of tumorsphere cultivation presents some limitations, such as differentiation and cell death, occurring in the sphere environment [112]. Alternatively, GSC enrichment has also been performed using adhesive cultures, which would provide uniform access to growth factors, thus avoiding differentiation [113, 114]. Phenotypic differences are observed within adhesive surfaces [115], such as in dishes previously treated with matrigel [116], laminin [113], collagen [117, 118] or extracellular matrix [82]. On the other hand, organotypic cultures in the presence of ECM and vascular elements are also available, especially for migratory assays, as this model represents a more accurate brain matrix micro-environment, in which cells migrate [119].
GSC enrichment performed in adhesive as well as non-adhesive cultures supports a major role for medium composition in this process [113, 118]. GSC culture medium, similar to that for NSCs, is supplemented with 20 ng/ml of EGF, 20 ng/ml of FGF-2 and sometimes with the 5 ng/ml of leukemia inhibitory factor (LIF). The mitogenes EGF and FGF are for long known for inducing renewal and expansion and notably increasing the frequency of spheres formed by undifferentiated cells [120, 121]. On the other hand, LIF has also been shown to enrich GSCs based on the transforming growth factor-beta (TGF-β)-mediated promotion of symmetric expansions inducing self-renewal and preventing differentiation [122], also increasing the expression of core stemness transcription factors and thus the number of NPCs [123]. Furthermore, medium supplemented with B27 and/or N2 enhances proliferation of multi-potent NPCs [124].
Interestingly, the basis behind the use of the defined medium is that self-renewal and multipotency of stem-like cells would be induced and sustained with a supplement medium switch from serum to EGF and FGF [125, 126]. EGF induces proliferation of adult mouse brain NSCs, generating spheres responsive to FGF-2. Then, upon stimulation by FGF-2, undifferentiated cells proliferate undergoing symmetric or asymmetric divisions and give rise to either unipotent (neuronal) or bipotent (neuronal/astroglial) progenitors [127]. The EGF-responsive mammalian embryonic neural precursor is a stem cell [121]. Later, other evidences supported that FGF-2 responsiveness was sufficient to isolate progenitors found in the adult mammalian spinal cord [128] and that NSCs responsive to FGF-2 arise earliest in embryonic development [129]. GSC marker expression was increased upon cultivation stimulation with FGF-2 compared to EGF [130]. In fact, blockade of EGF receptors reduced sphere growth, even in the absence of EGF in the culture medium. Both EGF and FGF enhanced GSC proliferation and sphere size. However, the absence of both mitogens in GBM culture was already demonstrated to originate multipotent spheres that were capable of self-renewal and also forming highly invasive tumors [131].
Lowering oxygen concentrations promotes GSC enrichment. Although the standard normoxic condition uses 20% of oxygen, this oxygen level is considered as hyperoxic compared to physiological brain oxygenation. In fact, hypoxia is the physiological environment of adult brain tissue, ranging from 1% to 5% of oxygen [132, 133]. Solid tumors including GBM are less oxygenated, ranging from physiological levels to below 0.1% in necrotic areas [134, 135]. Hypoxia regulates gene expression of hypoxia-inducible factor 1 (HIF-1), a transcription factor composed by an oxygen-regulated HIF-1α and a constitutive HIF-1β subunit. HIF-1α has a half-life of few minutes and destabilizes in increasing oxygen levels by the action of proline hydroxylase which utilize O2 in the range of 0.1 to 21% as a cofactor [136]. HIF post-translational modified within several domains interacts with the von Hippel-Lindau (pVHL) followed cytoplasmic translocation and ubiquinin-mediated for proteasome degradation [137, 138]. On the other hand, HIF-1α stabilized in hypoxic conditions translocates to the nucleus interacting with co-activators, such as cAMP, and binding to the hypoxia response element located (HRE) in the promoter or the enhancer regions of target genes [139]. To date, three HIF isoforms have been described, including HIF-2α and HIF-3α.
HIF-1α and HIF-2α expression was increased in GSCs exposed to hypoxic conditions together with other stem cell markers such as CD133, Bmi-1 and nestin [140]. Whereas HIF-1α is ubiquitously expressed, including by all hypoxic tumor cells, HIF-2α participates in cell reprogramming towards a cancer stem cell phenotype maintaining the GSC. HIF-1α target genes are regulated in a tissue-specific fashion. More than 100 downstream genes were identified as regulators of multiple physiological responses to oxygen deprivation, in general shifting the metabolic balance from oxidative phosphorylation toward glycolysis. HIF-1α is required for the proliferation, survival and angiogenesis [126]. Overstabilization of HIF-1α supports GBM resistance to TMZ [141]. Moreover, HIF-1α interaction with the activated Notch intracellular domain attenuates differentiation-promoting effects of bone morphogenetic proteins (BMPs), in particular of BMP-2. In NSCs and NPCs, increasing O2 levels degrade HIF-1α, thus promoting differentiation or apoptosis [142]. In GBM, knock down of HIF-1α at the RNA expression level reduced migration/invasion phenotypes beyond the ability to form tumorspheres [143]. A proposed mechanism is that HIF-1α drives the initial response to hypoxia whereas HIF-2 α drives the chronic response. The switch from HIF-1α- to HIF-2α-dependent signaling pathways plays divergent, however complementary roles during the hypoxic response reprograming GBM cells towards stemness, increasing aggressive tumor growth and invasiveness [144, 145]. However, HIF-2α stability is unaffected in physiological oxygen levels. Moreover, evidences support that HIF-2α is essential only by GSCs expressed and not by NPCs [146]. HIF-2α is overexpressed by GSCs acting as a mediator of tumor plasticity and tumorigenesis [126, 146]. A underlying mechanism could be that HIF-2α upregulates expression of stem-related genes such as Oct4 [147].
Remarkably, hypoxic microenvironment contributes to the GSC phenotype and increases the number of stem-like cells in tumor populations. Low 1% oxygen level enriches the percentage of CD133-expressing cells [148] without affecting N-glycosylation of this epitope [149]. A culture in hypoxia for 48 hours resulted in a four-times increase of the number of CD133+ cells [150]. Hypoxia-mediated HIF-1α activation results in a time-dependent increase in CD133 mRNA and protein levels with the peak of protein expression after 48 hours [149, 150]. The enrichment of GSCs from GBM primary cultures or the cell line U-87 under 1% oxygen condition results in tumorspheres smaller than the ones obtained in conditions of 20% oxygen. In agreement, expression of Ki-67, a marker of proliferation, is reduced in conditions of 1% oxygen, whereas expression of stemness markers, such as CD133, podoplanin, Bmi-1 and nestin, is increased. Sox-2 expression, in particular, is increased only in tumorspheres from primary cultures [140]. In terms of enrichment, 1% of oxygen increases the stem-like cells over fivefold with the percentage of CD133-positive cells being threefold or more enriched. In the same line, oxygen levels reduced to 7% enhance the stem-like phenotype of CD133-positive cells [151]. In addition to promoting proliferation, hypoxic conditions induced angiogenesis known to be a prerequisite of tumor expansion. Angiogenesis is favored by the association of GSCs and endothelial cells in perivascular niches [152]. Regulatory functions of such niche on GSCs is proposed in analogy to the ones in the control of NSC proliferation [153].
The effects of hypoxia on in vitro cell culture have gained scientific interest. Precise control of oxygen levels is essential for accurately interpreting obtained results. To date, hypoxic cell culture might be performed by different strategies. The most widely used incubator is based on a chamber flushed with 1% O2/5% CO2/94% N2 [154]. However, this chamber suffers frequently from the leakage of hypoxic conditions. As a main drawback of this approach, every time the incubator door is opened, a certain amount of oxygen needs to be consumed for re-establishing initial conditions. Alternatively, a hypoxic environment might be created by putting the cell dishes inside a subchamber with oxygen controller, which allows studies exploring multiple oxygen levels for each chamber to be placed inside a regular cell culture incubator. However, this alternative also implies in discontinuous oxygen levels and difficulties of reproducing exact hypoxic conditions.
For improving experimental conditions, hypoxic glove boxes emerged allowing cell manipulation without affecting oxygen levels. However, as the incubation and manipulation cells in the same humid station implies risk of contamination of cultures, the glove box station allowing hypoxic manipulation might be enclosed in a hypoxic incubator. Since exposure to room air also occurs, disrupting hypoxic condition during regular microscopy, a hypoxia microscope chamber should be added the workstation. In this sense, appropriate modular interconnected closed-hoods accommodating microscopes, centrifuges, cell sorters, bioreactors are receiving more attention as these ensure unprecedented stable and continuous hypoxia, completely isolated from the outside environment. In parallel, easy to use and low-cost alternatives have been developed, as, for instance, inflatable chambers made of transparent plastic materials allowing real-time studies of hypoxia under microscopy [155]. Finally, as the oxygen level is typically controlled in the gas phase differs from that supplied to the cells submerged in the growth medium; pericellular oxygen level monitoring might be used for better accuracy. This can be achieved with sensor chips embedded in a conventional tissue culture flask [156].
Vascularization is a diagnostic hallmark of GBM. At least five interlinked pathways by which GBM achieves neovascularization, have been described. A step of vascular co-option between brain and tumor vasculature, is followed by angiogenesis as second step. Vasculogenesis is the third mechanism sustained by the differentiation of circulating bone marrow-derived cells (BMDCs), known as endothelial progenitor cells (EPCs). These cells are suggested to promote a supportive role and might be incorporated into tumor vasculature. The fourth mechanism of neovascularization, termed vascular mimicry, is defined as the ability of tumor cells to form functional vessel-like networks. The fifth mechanism is based on the trans differentiation of GBM cells into an endothelial phenotype [157]. However, tumor cells involved in vascular mimicry are endowed with stemness plasticity, providing a source for transdifferentiation of GSCs towards the endothelial phenotype. The presence of GBM-derived vasculatures highlights the plasticity of GSCs, although the underlying mechanisms remain to be elucidated [158, 159].
Hypoxia is involved in all steps of the neovascularization process. HIF activation has been described to up-regulate VEGF expression and thus to promote angiogenesis [160], but also to increase levels of SDF-1, which in turn promote vasculogenesis recruiting CXCR4 positive BMDCs to the tumor site. However, unlike in normal vascularization, the emerging vasculature is often abnormal. In these pathological angiogenesis, the excess of vascular proliferation together with the lack of structure-giving pericytes contributes to the formation of tortuous and dilated blood vessels that are poorly organized and hyperpermeable (reviewed in [161]). Hence, vaso-occlusive and plasma coagulation impairs the blood supply perpetuating the hypoxic microenvironment, which in turn regulates HIF expression and activity thus leading to a vicious circle of induction of abnormal vasculature growth in GBM [162]. At the same time, hypoxic exosomes secreted by GBM cells induce endothelial cells to liberate growth factors and cytokines accelerating tumor growth [163].
In this context, GSCs are maintained within the vascular niche, in which the endothelial cells play an important role to sustain the GSC-like phenotype and GBM propagation. The endothelial secreted factors, which activate self-renewal pathways, including Hedgehog [164] and the Notch pathway [165, 166], at the same time sustain mTOR activation, which in turn suppresses autophagy and apoptosis of GSCs [167]. Both, endothelial cells and GSCs, express nestin and the cell surface antigen CD133 [168]. Almost 75% of nestin+ cells co-express CD133 while less than 0.1% of nestin+ cells coexpress the endothelial marker CD34 [116]. In addition, GBM SP cells, characterized by their efflux properties using flow cytometry were identified as brain endothelial cells [55].
Co-culture of GBM and endothelial cells results in a GSC population with higher proliferation rates and subsequently formation of bigger spheres and more aggressive tumors than observed with GSCs in monoculture [116]. On the other hand, co-culture of GSCs and endothelial cells also augments number and proliferation of endothelial cells [126]. The majority of GSC-enrichment studies employing co-culture conditions were done with immortalized human brain endothelial cells (hCMEC) [167, 169], umbilical vein endothelial cells (HUVEC) [164], microvascular endothelial cells (HMVEC) [126], primary endothelial cells [116] and primary bovine endothelial cells [117]. In general, trans-well inserts are used to co-culture endothelial and GSCs ensuring free diffusion of signaling molecules without direct cell contact [116, 117, 126, 164]. Alternatively, conditioned media from 72 h endothelial cell culture in serum-free medium might also be employed [167]. Endothelial cells cultivated in three-dimensional system enhanced secretion of IL-8, upregulating IL-8 cognate receptor CXCR1 and CXCR2 activities with the consequence of enhanced GSC migration, growth and stemness properties [169].
Pericytes are perivascular contractile stromal cells that surround the wall of endothelial cells regulating blood flow [170]. In GBM, it was demonstrated that pericytes mediate co-option of modified pre-existing blood vessels supporting the expansion of the tumor margin [161]. Besides, pericytes present mesenchymal stem cell (MSC) phenotypes, exhibiting neural multi potential activity [171, 172], also mediating immunosuppression effects on GBM. [173]. Interestingly, although GSCs have been suggested to differentiate into endothelial cells, recent evidence indicates that GSC preferentially differentiate into pericytes, attempting to support vasculature function and tumor growth [174, 175]. Besides, endothelial cells have been proposed to promote paracrine stimulation of pericytes, specifically in the context of hypoxia. In this context, hypoxic exosomes secreted by GBM cells enhance pericyte vessel coverage and induce endothelial cells to secrete factors stimulating pericyte PI3K/Akt pathway activation and migration [163]. Endosialin (CD248), not expressed by endothelial cells, is a marker of closely associated pericytes in GBM [176].
One of the initially described characteristics of GSCs is tumorigenicity in vivo [26]. Considering that GSCs present a distinct capacity for tumor growth and propagation, these cells have been enriched by serial passages in immune-compromised animals. The enrichment of human GSCs in vivo is based on tumor xeno-engrafting into immunosupressed animals (generally mice and rats). The congenitally athymic and hairless nude mouse immunodeficient in functional T cells has been routinely used in tumorigenesis assays and shown to be suitable for GSC transplantation due to its impaired capability of rejecting human xenografts tumors. On the other hand, the severe combined immunodeficient (SCID) mouse carries a genetic defect preventing functional development of T and B cells due to dysfunction in the TCR and BCR recombination machinery. However, the SCID mouse retains normal numbers of natural killer (NK) cells [177], which might interfere in the rejection of xenografts, as NK cells promote selective lysis of particular tumor targets [178]. An appropriate model for analysis of GSCs has been created by the inclusion of the non-obese diabetic background (NOD-SCID) with reduced NK cell function. Properly, injection of anti-NK cell antibodies before transplantation enhances the engraftment of human xenografts [179]. Followed by the NOD/SCID/γ cnull (NOG) strain, with the above-noted deficiencies including cytokine production incapability, the NOG strain has been considered a more appropriate animal recipients and especially suitable for GSC transplantation [180].
Intracranial GSC grafting has been widely performed using stereotactic approach, whereas few studies have explored subcutaneous or intraperitoneal engrafts. Engrafting cells into a mouse brain is a critical step as the site of injection determines the location of the tumor bulk and requires special care for in fact reaching the brain tissue [181]. Alternatively to stereotactic procedures, an implantable guide screw was developed to establish intracranial xenografts in mice, allowing a large number of animals to be engrafted [182]. Later, the guide screw method was modified incorporating an infusion pump that allows up to 10 animals to be simultaneously intracranial injected with tumor cells [181]. In general the number of intracranial injected cells was 105 cells per animal, although tumor formation was also observed with injection of 2 × 102 to 103 cells after 12 weeks [183]. Injection of 5 × 103 to 104 cells resulted in tumor establishment after 8 weeks. Tumor formation in vivo, a functional method for defining GSCs, is widely used since for enrichment of these cells. Cells are replicated by serial passages re-injecting them into secondary mice [26, 159], which also serve as a validation assay. Xenograft tumor excision later allows further histological and molecular characterization.
Tumor cells have less requirements for proliferation, in general being less independent from growth factor stimulation and anchorage when compared to normal cells. However, not every cell within the tumor present the same independence in order to survive and propagate. On the other hand, GSCs are considered to be capable of clonogenic proliferation as anchorage independent spheres under serum withdrawal, as similarly observed for NSCs. Also similar to NSCs, the frequency of GSCs has been predominantly assessed by primary spheres assays. The studies on GSCs usually refer to GBM primary tumors cultured with defined medium supplemented with EGF and FGF. Ignatova et al. [106] cultured tumor cells using non-adhesive dishes previously treated with poly-Hema and detected after 18 days a yield of GSC frequency from 0.05% to 1.26% (n = 10) in the GBM population. Moreover, the authors of this paper reported that the size of formed tumorspheres is heterogeneous (<50 cells or 50 to 2000 cells), and some tumors do not present no clonogenic cells at all. In another study published by Galli et al. [27], tumor cells cultured on adhesive surfaces for 20 to 40 days yield a GSC frequency of 0.5% to 31% (n = 12).
Another work reports that GSCs could by isolated following only 7 days of culture on adhesive surfaces with a yield of nearly 20% based on FACS separation of CD133-positive cells [184]. On the other hand, a subpopulation of 2–5% of CD133+ GSCs isolated by marker formed spheres after 42 days in culture whereas no sphere formation was observed in CD133− cells by Beier et al [104]. The work of Wang et al. (2010) agreed with the observation that CD133− cells were not capable of forming tumorspheres, while one of CD133+ cells actually contributed to tumorsphere formation. Another study resulted in the observation that small spheres (∼50 cells) were frequent and that 5% of plated cells were able to form secondary spheres, with around 70% of the sphere-forming cells being CD133+ [104]. Qiang et al. [105] confirmed that CD133+ cells presented higher colony-formation capacity including more G0/G1 phase cells.
GBM cell lines cultured using non-adhesive surfaces and presenting no ALDH activity (ALDH−) do not yield tumorspheres whereas GBM cell lines presenting ALDH activity (ALDH+) exhibit tumor sphere formation capacity [83]. A recent study supports this observation as ALDH+ cells isolated by FACS from primary GBM cultures on adhesive surfaces, previously treated with extracellular matrix, yield tumorspheres after only 1 day whereas ALDH− cells did not. Regarding hypoxic conditions, tumor cell culture in 1% of O2 for 5 days increased almost by three-fold the capacity of the tumor cells to form spheres [150]. Furthermore, hypoxia affects spheroid diameters and numbers forming tumorspheres. Sphere diameters were in average 50 μm in 1% O2 and 100 μm in 7%. However, hypoxia favored a higher proliferation rate [140]. On the other hand, conditioned media from immortalized human brain endothelial cells yield larger tumorspheres size [169].
Neurosphere assays for quantification of NSCs in vivo [185] remain an useful and low-cost tools for assessing GSC populations. However, the capacity to form tumorspheres has been criticized as a prediction for CSC enrichment [111]. Tumorsphere formation is not predictive for CSC expression markers, at least not for all types of cancer cells. Pastrana and co–workers critically evaluated the sphere-formation as an assay for stemness and pointed out that this assay may not detect quiescent stem cells. In contrast, the proposed assay detects cells with either tumorigenic properties in vivo or already dividing ones [186]. It is noteworthy that the source of multipotencial NSCs relies on a relatively quiescent cell and not on the proliferating population [187]. For the determination of self-renewal capacity, cultures should be plated as single cells to ensure that tumorsphere formation is only due to truly clonogenic proliferation instead of cell fusion and/or aggregation, which might occur even at very low culture density. On the other hand, as cell survival and proliferation are directly affected by paracrine stimulation, very low culture density conditions suffer from poor dispersion of signaling molecules in the medium [186]. In this case, a good option to improve the sphere-formation assay is to plate cells with previously conditioned and filtered medium. Additionally, a software specially suitable for comparing populations depleted and enriched in stem cells is available [188].
GSCs are usually functionally defined by their tumorigenic property in immunosupressed animals. As self-renewal is a key feature of stemness, GSC identity relies on serial transplantation capability. Singh and co-workers showed that after 5 weeks of reinjection of 1,000 CD133+ cells in NOD-SCID mouse brains (n = 3) the generated tumors recapitulated the phenotype of the original patient primary tumor whereas CD133− cells took 12 weeks after transplantation to exhibit tumor formation [26]. Of note, the first sign of neurologic impairment in mice was observed 8–10 weeks after transplantation of GSCs. Galli and co-workers observed that GSCs gave rise to tumors that mimic GBM, which did not occur in the U-87 MG cell line. Moreover, GSCs presented migratory capacity typical for GBM, which was also not observed with U-87 MG cells [27]. In contrast, Pollard et al. [113] demonstrated tumor engraftment using a minimum number of 100 cells, whose isolation was not based on CD133 expression, but on exploring adhesive culturing. Indeed, Qiang et al. pointed out that seven weeks of culture on non-adhesive surfaces generated larger subcutaneous tumor xenografts than compared to those obtained in cultures on adhesive surfaces [105]. Compared with CD133+ cells enriched in the presence of primary endothelial cells, xenografts were generated within 7 days of injection of 1,000,000 cells [116]. Co-culture with endothelial cells increases tumor formation by a factor of six compared to the monoculture. After 11 weeks, animals intracranially injected with 100,000 cells formed tumors with a size of 110 mm3, whereas previous co-cultures with endothelial cells resulted in 600 mm3 tumors [169]. In mice, co-injection of GL261glioma and b.END3 endothelial cells resulted in larger allografts when those observed following transplantation of glioma cells alone [164].
Alternative methods for animal experimentation include GBM growth in semi-solid medium employing agar or agarose suspension [189, 190]. Agarose-suspension culture is for long known to be one of the best and low cost in vitro assays correlating with tumorigenicity [108, 189, 190], still used for GSC validation [191–193]. Alternatively, GSC enrichment has also been performed using agarose as non-adhesive surface [194].
GSCs can be validated based on expression patterns of classical NSC markers, correlating with the progression of WHO grades astrocytomas [195]. Among them, the transcription factor Octamer 4 (OCT-4), a marker of stem cell pluripotency [196], has been employed. OCT-3/4 promotes migration and invasion of GBM [197], whose knockdown was described to result in CSC apoptosis [114]. In stem cells, OCT-4 expression was shown to be upregulated by HIF-2α during hypoxia, increasing its mRNA level four times [150]. However, in another studied OCT-4 expression could not be detected in GSC, although performed with GSC enriched in adhesive cultures previously treated with laminin [126]. On the other hand, SRY (sex determining region Y)-box 2, also known as SOX-2, which is a encoding group of transcription factors, is also found in NPCs and are also used as GSC validation marker [83]. Musashi encodes for various RNA-binding proteins which contribute to the maintenance of stemness during CNS development and are also applied in the validation of the GSC phenotype [83, 150]. Nestin is an intermediate filament protein specific for glia, usually applied as a multipotent marker in NSC characterization and also in the validation of the GSC phenotype [198]. Nestin-knockout embryos revealed reduced self-renewal with no overt defects in cell proliferation or differentiation, surprisingly uncoupled from nestin's structural involvement in the cytoskeleton [199]. Nestin has been found to be highly expressed in invasive GBM xenografts delineating tumor infiltration [200]. However, as neurogenesis precedes gliogenesis during development, the more undifferentiated stem cell-like phenotypes are thus negative for nestin expression and found nestin+ later [112]. In line with this observation, nestin− mouse NPCs gave raise to neurons and glia [201]. In addition, 75% of nestin+ cells in GBM coexpressed CD133 [116]. However, in GSCs after 5 days of differentiation with serum, nestin expression levels were still at a high level, whereas CD133 was much lower expressed [105]. CD133 is the most used marker for GSC phenotype validation. However, a controversial discussion remains about the suitability of CD133 expression for validating and targeting GSCs. Beier and co-workers supported evidences that CD133+ GSCs maintain only a subset of primary GBMs [104]. At the same time, CD133− derived GBM cells formed tumors in nude rats beyond the efficiency of CD133+ cells [61]. In addition, CD133 has been presented as marker of bioenergetic stress in GBM rather than for being a marker protein of their stem-like properties [202]. However, considering that surface phenotype of stem cells may remain intact despite decay in functional activity, a more functional assay would be appropriate, as those employing ALDH expression. According to Choi and co-workers ALDH+ cells range from 0.3–28.9% in GBM primary cultures. Furthermore, near 40% of ALDH+ cells overlapped with CD133 expression whereas only near 0.3% of CD133+ cells exhibit ALDH activity [82].
GSCs are proposed to be chemoresistant, a feature explicating GBM recurrence after conventional therapy [38]. Attempting to predict this chemoresistant phenotype, in vitro chemotherapeutic drug assays assess the chemosensitivity of CSCs [194]. Temozolomide (TMZ) is a chemotherapeutic alkylating agent included in the standard therapy against GBM. Although experimental data demonstrate that TMZ preferentially depletes GSCs [203], and that primary GBM cell response to therapy occurs in patient-specific fashion and independent of GSC phenotype [204], the majority of studies regarding the susceptibility of GSCs points at a chemoresistant phenotype of GSCs towards alkylating agents [205]. TMZ induces apoptosis and senescence in GBM cells cultured as tumorspheres, a phenotype also used for GSC validation [206]. In fact, conversion of differentiated GBM cells into GSCs could take place upon treatment with TMZ [207].
Recently, TMZ was described to down-regulate P-glycoprotein expression, subsequently promoting survival of GSCs by the Wnt3a/glycogen synthase-3 kinase/β-catenin signaling pathway [208]. In line with this observation, BMP2-induced differentiation sensitized GSC to TMZ therapy [141]. However, GSCs are not uniformly resistant to TMZ. In fact, CSCs are neither resistant nor susceptible to chemotherapy per se. DNA repair mechanisms restore the integrity of alkylated DNA bases thus contributing to drug resistance and subsequent tumor reoccurrence.
O6-methylguanine-DNA-methyltransferase (MGMT), a DNA repair enzyme, possesses detoxifying functions during TMZ treatment, when the MGMT gene is non-methylated, conferring a strong intrinsic resistance to GSC [209]. However, MGMT methylatio stauts alone sill does not predict the TMZ response with high precision as atypical TMZ-resistant GSC presented both methylated and unmethylated forms [210]. TMZ resistance of GSCs via regulation of MGMT expression is promoted by c-Jun N-terminal kinase (JNK) and Ras-Raf-MEK-ERK signaling pathways, whose inhibition enhances cytotoxicity of TMZ on GBM [211–213]. In addition to MGMT, another DNA repair protein, ALKBH2, was shown to mediate TMZ resistance in human GBM [214], however its specific effects on GSCs remain unknown. Recently, microRNA-125b has been shown to play a role in the resistance of GSCs against TMZ through down regulation of Bak1 and PIAS3 (protein inhibitor of activated STAT3) expression [215, 216]. In view of that, detection of microRNA-125b expression levels may be useful for validation of the GSC phenotype.
GSCs are postulated to recapitulate the heterogeneity of the parental tumor in immunosupressed animals due to their multipotency capacity. As the differentiation property is a key feature of stemness, differentiation assays attempt to evaluate the functional GSC phenotype. Similar to assessing stemness in NSCs, the assay is done by cultivating GSC under pro-differentiation conditions. Thus, upon induction of differentiation, cells are going to express specific genes and proteins of the three cell types of neural lineage (neurons, oligodendrocytes and astrocytes). These results suggest that initial cells have stem cell-like characteristics in vitro. Differentiation progress is usually evaluated based on expression determination of cytoskeleton-associated proteins such as neuron-specific β3-tubulin (TuJ1) and MAP2, or glial cell-specific GFAP [217]. Moreover, glycoproteins, postulated to determine stem cell fates, are differentially expressed during differentiation, and used to determine the differentiation status of GSCs [218]. The loss of the CD133 epitope, ABC transporters, and ALDH-1 also are evidence for CSC differentiation [56, 60, 73].
The glial differentiation of GSCs, evidenced by increased expression of GFAP, can be induced by all-trans retinoic acid (RA) GSC cultures were cultured in growth factor-free medium [70]. Similarly, BMPs promote also astro-glial differentiation of GSCs [219]. Interestingly, BMPs were already demonstrated to sensitize GSC for TMZ by affecting HIF-1α stability and MGMT expression [141]. Lee et al. [217] showed that GBM-derived GSCs cultured with N2 supplement, RA or 10% fetal bovine serum alone, within a month progressively differentiate losing their NSC markers nestin, SSEA-1 and Sox2 and developing morphologies and immunohistochemical staining patterns (GFAP, TuJ1 and MAP2), consistent with phenotypes of glial and neuronal lineages. Finally, in vivo experiments with immunosupressed animals provide a proper environment for differentiation of GSCs, recapitulating GBM mass with a variety of cell types [26]. Therefore, since differentiation property is a key feature of stemness, this process might be applied in the validation of GSC phenotypes.
A number of methods have been used to identify GSCs. Emerging evidence suggests that GSCs do not comprise a fixed entity, but a state-dependent phenotype of the micro-environmental niche. In this sense, diverging strategies to obtain GSC hinders inter-laboratory comparisons when not giving rise to conflicting results. Enrichment and isolation methods proposed to obtain GSC are evolving, allowing mimicking in vivo niches to enrich GSC culturing in vitro and introducing novel markers for cytometry isolation. Hypoxic and co-cultivation with endothelial cells enriches the cell population with the expected GSC phenotype. In line with this, the isolation of GSCs based on expression of CD133 remains a valuable approach, while ALDH expression has turned into a promising strategy for GSC isolation, contemplating functional and viable assessment of the GSC phenotype (see Figure 1 for a comprehensive scheme). On the other hand, validation assays are usually performed by measuring the capacity of forming tumorspheres, stemness marker expression and tumorigenesis in vivo. However, these strategies for validation might overlap with those used to enrich and/or isolate GSCs, such as in the endless möbius ribbon paradigm. Similar to this paradigm, the overlap of the methods of enrichment/isolation with the ones to validate the obtained GSC is presented in an endless cycle potentially introducing bias into the process to obtain bona fide GSC. In this sense, differentiation assays, together with chemo-resistance assays, assess more interesting predictive functional features of GSC, although precautions regarding the expression of DNA repair genes should be taken in account in order to interprete the latter test. Remarkably, little knowledge exists on quiescent GSCs, the subset of GBM cells responsible for the production of transient populations of highly proliferative cells. Notably, the hierarchy of markers during NSC versus NPC stage transition remains obscure, especially considering the high plasticity of this process, with more committed NPCs reverting back to a more primitive NSC state. Interestingly, ALDH expression has been suggested as marker for distinguishing between NSCs and NPCs. On the other hand, the sphere frequency assay is more suitable for progenitor cell than for stem cell activity. In summary, the scheme presented in Figure 2 puts together how methods used for GSC enrichment and isolation might interfere with the parameters used for GSC phenotype validation. Investigation of GSC as a relatively recent field relies on the establishment and optimization of universal methods focusing at isolation of GSC and robust multi-parameter characterization
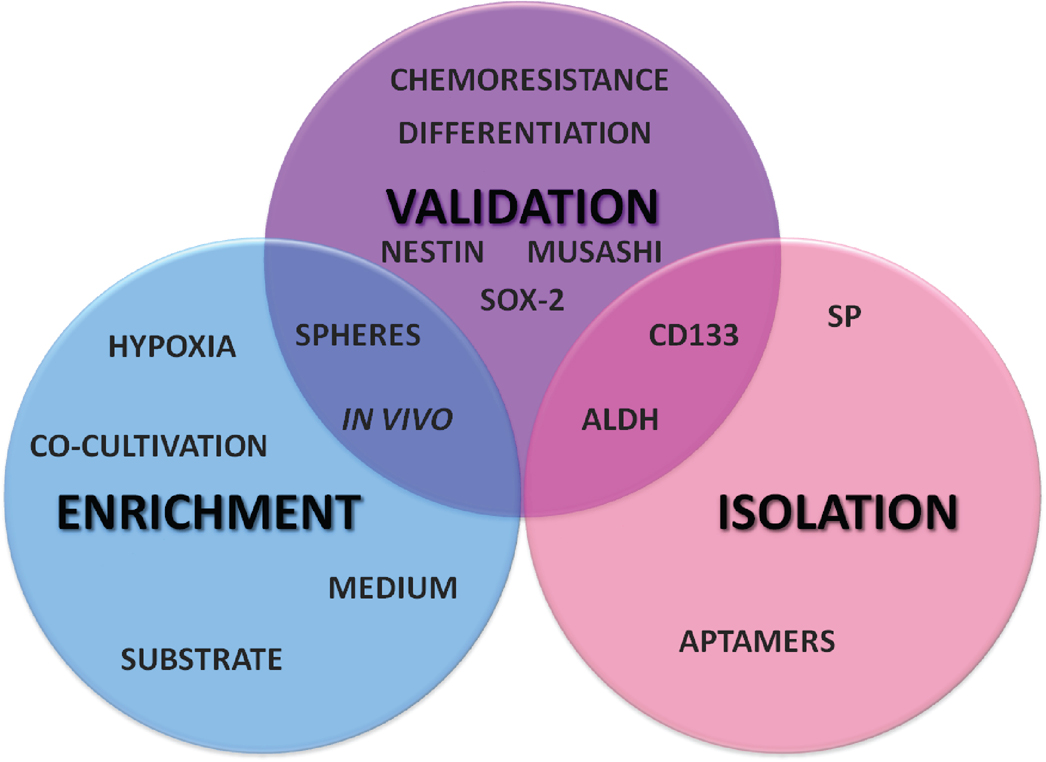 |
Figure 2. Multi-parameter panel for GSC enrichment, isolation and validation. The GSC panel summarizes usually employed methods for isolation and enrichment of GSCs compared to the ones for GSC-phenotype validation. GSC assessment usually takes into account differential biomarker expression, sphere formation and tumorigenesis in vivo capacities, which might overlap with the parameters of the chosen methods for GSC enrichment and/or isolation methods. Robust GSC validation assays include other functional aspects, such as chemoresistance and multipotent differentiation capacities. |
HU acknowledges grant support from FAPESP and CNPq, Brazil. TLS acknowledges Slovenian Agency for Research (ARRS) for supporting the work by granting the Program P1-0105-0245 to TTL. TLS is grateful for a visiting professor fellowship granted by the Sciences without Frontiers Program of the CNPq. ESM and MPP are grateful for fellowship support by the Brazilian funding agencies FAPESP and CNPq, respectively.
| [1] | Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007, 114:97–109. |
| [2] | Ohgaki H, Burger P, Kleihues P. Definition of primary and secondary glioblastoma–response. Clin Cancer Res 2014, 20:2013. |
| [3] | Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ, Norden AD, et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 2010, 60:166–193. |
| [4] | Altaner C, Altanerova V. Stem cell based glioblastoma gene therapy. Neoplasma 2012, 59:756–760. |
| [5] | Lima FR, Kahn SA, Soletti RC, Biasoli D, Alves T, da Fonseca AC, Garcia C, Romão L, Brito J, Holanda-Afonso R, Faria J, Borges H, Moura-Neto V. Glioblastoma: therapeutic challenges, what lies ahead. Biochim Biophys Acta 2012, 1826:338–349. |
| [6] | The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455:1061–1068. |
| [7] | Masui K, Cloughesy TF, Mischel PS. Review: molecular pathology in adult high-grade gliomas: from molecular diagnostics to target therapies. Neuropathol Appl Neurobiol 2012, 38:271–291. |
| [8] | Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, et al. TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell 2013, 155:462–477. |
| [9] | Riddick G, Fine HA. Integration and analysis of genome-scale data from gliomas. Nat Rev Neurol 2011, 7:439–450. |
| [10] | Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005, 352:997–1003. |
| [11] | Kahn SA, Biasoli D, Garcia C, Geraldo LH, Pontes B, Sobrinho M, Frauches AC, Romão L, Soletti RC, et al. Equinatoxin II potentiates temozolomide- and etoposide-induced glioblastoma cell death. Curr Top Med Chem 2012, 12:2082–2093. |
| [12] | O'Connor ML, Xiang D, Shigdar S, Macdonald J, Li Y, Wang T, Pu C, Wang Z, Qiao L, Duan W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett 2014, 344:180–187. |
| [13] | Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011, 144:646–674. |
| [14] | Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, Chomienne C, Ishikawa F, Schuringa JJ, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer 2012, 12:767–775. |
| [15] | Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea - a paradigm shift. Cancer Res 2006, 66:1883–1890. |
| [16] | Alison MR, Lim SML, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol 2011, 223:147–61. |
| [17] | Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001, 414:105–111. |
| [18] | Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008, 8:755–768. |
| [19] | Shiozawa Y, Nie B, Pienta KJ, Morgan TM, Taichman RS. Cancer stem cells and their role in metastasis. Pharmacol Ther 2013, 138:285–293. |
| [20] | Chaffer CL, Brueckmann I, Scheel C, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011, 108:7950–7955. |
| [21] | Li Y, Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res 2012, 72:576–580. |
| [22] | Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol 2013, 15:338–344. |
| [23] | Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJA. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer 2005, 5:899–904. |
| [24] | Prestegarden L, Enger PØ. Cancer stem cells in the central nervous system–a critical review. Cancer Res 2010, 70:8255–8258. |
| [25] | Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res 2003, 63:5821–5828. |
| [26] | Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature 2004, 432:396–401. |
| [27] | Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004, 64:7011–7021. |
| [28] | Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, Greve JM, Soriano RH, Gilmour LL, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17:362–375. |
| [29] | Tajnšek U, Motaln H, Levicar N, Rotter A, Lah TT. The duality of stem cell: double-edged sword in tumour evolution and treatment. In: Resende RR, Ulrich H, editors. Trends in Stem Cell Proliferation and Cancer Research. Dordrecht: Springer Netherlands, 2013:391–434. |
| [30] | Chen C, Chai J, Singh L, Kuo CY, Jin L, Feng T, Marzano S, Galeni S, Zhang N, et al. Characterization of an in vitro differentiation assay for pancreatic-like cell development from murine embryonic stem cells: detailed gene expression analysis. Assay Drug Dev Technol 2011, 9:403–419. |
| [31] | Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17:98–110. |
| [32] | Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9:157–173. |
| [33] | Mantamadiotis T, Taraviras S. Self-renewal mechanisms in neural cancer stem cells. Front Biosci (Landmark Ed) 2010, 16:598–607. |
| [34] | Zheng S, Fu J, Vegesna R, Mao Y, Heathcock LE, Torres-Garcia W, Ezhilarasan R, Wang S, McKenna A, et al. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev 2013, 27:1462–1472. |
| [35] | Wan F, Herold-Mende C, Campos B, Centner FS, Dictus C, Becker N, Devens F, Mogler C, Felsberg J, et al. Association of stem cell-related markers and survival in astrocytic gliomas. Biomarkers 2011, 16:136–143. |
| [36] | Campos B, Zeng L, Daotrong PH, et al. Expression and regulation of AC133 and CD133 in glioblastoma. Glia 2011, 59:1974–1986. |
| [37] | Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008, 26:2839–2845. |
| [38] | Chen J, Li Y, Yu T-S, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488:522–526. |
| [39] | Cho DY, Lin SZ, Yang WK, Lee HC, Hsu DM, Lin HL, Chen CC, Liu CL, Lee WY, Ho LH. Targeting cancer stem cells for treatment of glioblastoma multiforme. Cell Transplant 2013, 22:731–739. |
| [40] | Rahman M, Deleyrolle L, Vedam-Mai V, Azari H, Abd-El-Barr M, Reynolds BA. The cancer stem cell hypothesis: failures and pitfalls. Neurosurgery 2011, 68:531–545, discussion 545. |
| [41] | Yu SC, Ping YF, Yi L, Zhou ZH, Chen JH, Yao XH, Gao L, Wang JM, Bian XW. Isolation and characterization of cancer stem cells from a human glioblastoma cell line U87. Cancer Lett 2008, 265:124–134. |
| [42] | Chaffer CL, Weinberg RA. Cancer cell of origin: spotlight on luminal progenitors. Cell Stem Cell 2010, 7:271–272. |
| [43] | Greve B, Kelsch R, Spaniol K, Eich HT, Götte M. Flow cytometry in cancer stem cell analysis and separation. Cytom A 2012, 81:284–293. |
| [44] | Watanabe M, Uehara Y, Yamashita N, Fujimura Y, Nishio K, Sawada T, Takeda K, Koizumi F, Koh Y. Multicolor detection of rare tumor cells in blood using a novel flow cytometry-based system. Cytom A 2014, 85:206–213. |
| [45] | Ulrich H, Tárnok A. Flow cytometry detection of circulating tumor cells: achievements and limitations as prognostic parameters. Cytom A 2014, 85:201–202. |
| [46] | Van Hoof D, Lomas W, Hanley MB, Park E. Simultaneous flow cytometric analysis of IFN-γ and CD4 mRNA and protein expression kinetics in human peripheral blood mononuclear cells during activation. Cytom A 2014, 85:894–900. |
| [47] | Markovic S, Li B, Pera V, et al. A computer vision approach to rare cell in vivo fluorescence flow cytometry. Cytom A 2013, 83:1113–1123. |
| [48] | Galanzha EI, Zharov VP. Circulating Tumor Cell Detection and Capture by Photoacoustic Flow Cytometry in vivo and ex Vivo. Cancers (Basel) 2013, 5:1691–1738. |
| [49] | Patrawala L, Calhoun T, Schneider-Broussard R, et al. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res 2005, 65:6207–6219. |
| [50] | Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A 2004, 101:14228–14233. |
| [51] | Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A 2004, 101:781–786. |
| [52] | Fukaya R, Ohta S, Yamaguchi M, Fujii H, Kawakami Y, Kawase T, Toda M. Isolation of cancer stem-like cells from a side population of a human glioblastoma cell line, SK-MG-1. Cancer Lett 2010, 291:150–157. |
| [53] | Weber K, Paulus W, Senner V. The side population of gliomas exhibits decreased cell migration. J Neuropathol Exp Neurol 2010, 69:623–631. |
| [54] | Broadley KW, Hunn MK, Farrand KJ, Price KM, Grasso C, Miller RJ, Hermans IF, McConnell MJ. Side population is not necessary or sufficient for a cancer stem cell phenotype in glioblastoma multiforme. Stem Cells 2011, 29:452–461. |
| [55] | Golebiewska A, Bougnaud S, Stieber D, Brons NH, Vallar L, Hertel F, Klink B, Schröck E, Bjerkvig R, et al. Side population in human glioblastoma is non-tumorigenic and characterizes brain endothelial cells. Brain 2013, 136:1462–1475. |
| [56] | Rama AR, Alvarez PJ, Madeddu R, Aranega A. ABC transporters as differentiation markers in glioblastoma cells. Mol Biol Rep 2014, 41:4847–4851. |
| [57] | Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006, 5:67. |
| [58] | Oliveira SL, Pillat MM, Cheffer A, Lameu C, Schwindt TT, Ulrich H. Functions of neurotrophins and growth factors in neurogenesis and brain repair. Cytom A 2013, 83:76–89. |
| [59] | Campos B, Herold-Mende CC. Insight into the complex regulation of CD133 in glioma. Int J Cancer 2011, 128:501–510. |
| [60] | Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, Zeilstra J, Pals ST, Mehmet H, et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res 2010, 70:719–729. |
| [61] | Wang J, Sakariassen PØ, Tsinkalovsky O, Immervoll H, BØe SO, Svendsen A, Prestegarden L, RØsland G, Thorsen F, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer 2008, 122:761–768. |
| [62] | Crea F, Fornaro L, Masi G, Falcone A, Danesi R, Farrar W. Faithful markers of circulating cancer stem cells: is CD133 sufficient for validation in clinics? J Clin Oncol 2011, 29:3487–3488, author reply 3488–3490. |
| [63] | Vasiliou V, Bairoch A, Tipton KF, Nebert DW. Eukaryotic aldehyde dehydrogenase (ALDH) genes: human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics 1999, 9:421–434. |
| [64] | Hilton J. Role of aldehyde dehydrogenase in cyclophosphamide-resistant L1210 leukemia. Cancer Res 1984, 44:5156–5160. |
| [65] | Parajuli B, Georgiadis TM, Fishel ML, Hurley TD. Development of selective inhibitors for human aldehyde dehydrogenase 3A1 (ALDH3A1) for the enhancement of cyclophosphamide cytotoxicity. Chembiochem 2014, 15:701–712. |
| [66] | Mimeault M, Batra SK. Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Mol Asp Med 2013, 39:1–2. |
| [67] | Schäfer A, Teufel J, Ringel F, Bettstetter M, Hoepner I, Rasper M, Gempt J, Koeritzer J, Schmidt-Graf F, et al. Aldehyde dehydrogenase 1A1–a new mediator of resistance to temozolomide in glioblastoma. Neuro Oncol 2012, 14:1452–1464. |
| [68] | Liu P, Brown S, Channathodiyil P, Kannappan V, Armesilla AL, Darling JL, Wang W. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer 2012, 107:1488–1497. |
| [69] | Yoshida A, Hsu LC, Davé V. Retinal oxidation activity and biological role of human cytosolic aldehyde dehydrogenase. Enzyme 1992, 46:239–244. |
| [70] | Shi Z, Lou M, Zhao Y, Zhang Q, Cui D, Wang K. Effect of all-trans retinoic acid on the differentiation of U87 glioma stem/progenitor cells. Cell Mol Neurobiol 2013, 33:943–951. |
| [71] | Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1:555–567. |
| [72] | Jiang H, White EJ, Conrad C, Gomez-Manzano C, Fueyo J. Autophagy pathways in glioblastoma. Methods Enzymol 2009, 453:273–286. |
| [73] | Moreb JS. Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther 2008, 3:237–246. |
| [74] | Soehngen E, Schaefer A, Koeritzer J, Huelsmeyer V, Zimmer C, Ringel F, Gempt J, Schlegel J. Hypoxia upregulates aldehyde dehydrogenase isoform 1 (ALDH1) expression and induces functional stem cell characteristics in human glioblastoma cells. Brain Tumor Pathol 2013, 31:247–256. |
| [75] | Adam SA, Schnell O, Pöschl J, Eigenbrod S, Kretzschmar HA, Tonn JC, Schüller U. ALDH1A1 is a marker of astrocytic differentiation during brain development and correlates with better survival in glioblastoma patients. Brain Pathol 2012, 22:788–797. |
| [76] | Jones RJ, Barber JP, Vala MS, Collector MI, Kaufmann SH, Ludeman SM, Colvin OM, Hilton J. Assessment of aldehyde dehydrogenase in viable cells. Blood 1995, 85:2742–2746. |
| [77] | Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci U S A 1999, 96:9118–9123. |
| [78] | StemCell Technologies. Technical bulletin 2009, 33:4–7. |
| [79] | Caldera V, Mellai M, Annovazzi L, Monzeglio O, Piazzi A, Schiffer D. MGMT hypermethylation and MDR system in glioblastoma cancer stem cells. Cancer Genomics Proteomics 2012, 9:171–178. |
| [80] | Smith CA, Colvin M, Storms RW, Ludeman SM. BODIPY aminoacetaldehyde diethyl acetal 2010. http://www.google.com/patents/EP1975243B1?cl=en. |
| [81] | Corti S, Locatelli F, Papadimitriou D, Donadoni C, Salani S, Del Bo R, Strazzer S, Bresolin N, Comi GP. Identification of a primitive brain-derived neural stem cell population based on aldehyde dehydrogenase activity. Stem Cells 2006, 24:975–985. |
| [82] | Choi SA, Lee JY, Phi JH, Wang KC, Park CK, Park SH, Kim SK. Identification of brain tumour initiating cells using the stem cell marker aldehyde dehydrogenase. Eur J Cancer 2014, 50:137–149. |
| [83] | Schäfer A, Teufel J, Ringel F, Bettstetter M, Hoepner I, Rasper M, Gempt J, Koeritzer J, Schmidt-Graf F, et al. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro Oncol 2010, 12:1024–1033. |
| [84] | Almanaa TN, Geusz ME, Jamasbi RJ. A new method for identifying stem-like cells in esophageal cancer cell lines. J Cancer 2013, 4:536–548. |
| [85] | Ueda K, Ogasawara S, Akiba J, Nakayama M, Todoroki K, Ueda K, Sanada S, Suekane S, Noguchi M, et al. Aldehyde dehydrogenase 1 identifies cells with cancer stem cell-like properties in a human renal cell carcinoma cell line. PLoS One 2013, 8:e75463. |
| [86] | Moreb JS, Ucar D, Han S, Amory JK, Goldstein AS, Ostmark B, Chang LJ, Moreb JS, Ucar D, et al. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem Biol Interact 2012, 195:52–60. |
| [87] | Naujokat C. Monoclonal antibodies against human cancer stem cells. Immunotherapy 2014, 6:290–308. |
| [88] | Ulrich H, Martins AHB, Pesquero JB. RNA and DNA aptamers in cytomics analysis. Cytom A 2004, 59:220–231. |
| [89] | Majumder P, Trujillo CA, Lopes CG, Resende RR, Gomes KN, Yuahasi KK, Britto LR, Ulrich H. New insights into purinergic receptor signaling in neuronal differentiation, neuroprotection, and brain disorders. Purinergic Signal 2007, 3:317–331. |
| [90] | Šmuc T, Ahn I-Y, Ulrich H. Nucleic acid aptamers as high affinity ligands in biotechnology and biosensorics. J Pharm Biomed Anal 2013, 81–82:210–217. |
| [91] | Ulrich H, Wrenger C. Disease-specific biomarker discovery by aptamers. Cytom A 2009, 75:727–733. |
| [92] | Kang D, Wang J, Zhang W, Song Y, Li X, Zou Y, Zhu M, Zhu Z, Chen F, Yang CJ. Selection of DNA aptamers against glioblastoma cells with high affinity and specificity. PLoS One 2012, 7:e42731. |
| [93] | Tan Y, Shi YS, Wu XD, Liang HY, Gao YB, Li SJ, Zhang XM, Wang F, Gao TM. DNA aptamers that target human glioblastoma multiforme cells overexpressing epidermal growth factor receptor variant III in vitro. Acta Pharmacol Sin 2013, 34:1491–1498. |
| [94] | Wu X, Liang H, Tan Y, Yuan C, Li S, Li X, Li G, Shi Y, Zhang X. Cell-SELEX aptamer for highly specific radionuclide molecular imaging of glioblastoma in vivo. PLoS One 2014, 9:e90752. |
| [95] | Shigdar S, Qiao L, Zhou SF, Xiang D, Wang T, Li Y, Lim LY, Kong L, Li L, Duan W. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett 2013, 330:84–95. |
| [96] | Kim Y, Wu Q, Hamerlik P, Hitomi M, Sloan AE, Barnett GH, Weil RJ, Leahy P, Hjelmeland AB, Rich JN. Aptamer identification of brain tumor initiating cells. Cancer Res 2013, 73:4923–4936. |
| [97] | Gao H, Qian J, Yang Z, Pang Z, Xi Z, Cao S, Wang Y, Pan S, Zhang S, et al. Precise glioma targeting of and penetration by aptamer and peptide dual-functioned nanoparticles. Biomaterials 2012, 33:5115–5123. |
| [98] | Dilnawaz F, Singh A, Mewar S, Sharma U, Jagannathan NR, Sahoo SK. The transport of non-surfactant based paclitaxel loaded magnetic nanoparticles across the blood brain barrier in a rat model. Biomaterials 2012, 33:2936–2951. |
| [99] | Huang YF, Shangguan D, Liu H, Phillips JA, Zhang X, Chen Y, Tan W. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem 2009, 10:862–868. |
| [100] | Yang T, Wang Y, Li Z, Dai W, Yin J, Liang L, Ying X, Zhou S, Wang J, et al. Targeted delivery of a combination therapy consisting of combretastatin A4 and low-dose doxorubicin against tumor neovasculature. Nanomedicine 2012, 8:81–92. |
| [101] | Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255:1707–1710. |
| [102] | Reynolds B, Rietze R. Neural stem cells and neurospheres—re-evaluating the relationship. Nat Methods 2005, 2:333–336. |
| [103] | Hong X, Chedid K, Kalkanis SN. Glioblastoma cell line-derived spheres in serum-containing medium versus serum-free medium: a comparison of cancer stem cell properties. Int J Oncol 2012, 41:1693–1700. |
| [104] | Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 2007, 67:4010–4015. |
| [105] | Qiang L, Yang Y, Ma YJ, Chen FH, Zhang LB, Liu W, Qi Q, Lu N, Tao L, et al. Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett 2009, 279:13–21. |
| [106] | Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39:193–206. |
| [107] | Ledur PF, Villodre ES, Paulus R, Cruz LA, Flores DG, Lenz G. Extracellular ATP reduces tumor sphere growth and cancer stem cell population in glioblastoma cells. Purinergic Signal 2012, 8:39–48. |
| [108] | Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell 1974, 3:355–359. |
| [109] | Yang MY, Chiao MT, Lee HT, Chen CM, Yang YC, Shen CC, Ma HI. An innovative three-dimensional gelatin foam culture system for improved study of glioblastoma stem cell behavior. J Biomed Mater Res B Appl Biomater 2014. |
| [110] | Steadman K, Stein WD, Litman T, Yang SX, Abu-Asab M, Dutcher SK, Bates S. PolyHEMA spheroids are an inadequate model for the drug resistance of the intractable solid tumors. Cell Cycle 2008, 7:818–829. |
| [111] | Calvet CY, André FM, Mir LM. The culture of cancer cell lines as tumorspheres does not systematically result in cancer stem cell enrichment. PLoS One 2014, 9:e89644. |
| [112] | Suslov ON, Kukekov VG, Ignatova TN, Steindler DA. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci U S A 2002, 99:14506–14511. |
| [113] | Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, Squire JA, Smith A, Dirks P. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4:568–580. |
| [114] | Hu T, Liu S, Breiter DR, Wang F, Tang Y, Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res 2008, 68:6533–6540. |
| [115] | Kamble SC, Bapat SA. Stem cell and cancer stem cell games on the ECM field. J Cancer Stem Cell Res 2013, 1:1–16. |
| [116] | D Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11:69–82. |
| [117] | Anfuso CD, Motta C, Giurdanella G, Arena V, Alberghina M, Lupo G. Endothelial PKCα-MAPK/ERK-phospholipase A2 pathway activation as a response of glioma in a triple culture model. A new role for pericytes? Biochimie 2014, 99:77–87. |
| [118] | Motegi H, Kamoshima Y, Terasaka S, Kobayashi H, Houkin K. A novel adherent culture method of glioblastoma cells expressing CD133 using collagen-1-coated plates. Hokkaido Igaku Zasshi 2012, 87:147–151. |
| [119] | Guerreto-Cázares H, Chaichana KL, Quiñones-Hinojosa A. Cancer Stem Cells. Methods Mol Biol 2009, 568:9–11. |
| [120] | Nieto-Estévez V, Pignatelli J, Araúzo-Bravo MJ, Hurtado-Chong A, Vicario-Abejón C. A global transcriptome analysis reveals molecular hallmarks of neural stem cell death, survival, and differentiation in response to partial FGF2 and EGF deprivation. PLoS One 2013, 8:e53594. |
| [121] | Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 1996, 175:1–13. |
| [122] | Peñuelas S, Anido J, Prieto-Sánchez RM, Folch G, Barba I, Cuartas I, García-Dorado D, Poca MA, Sahuquillo J, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009, 15:315–327. |
| [123] | Buono K, Vadlamuri D. Leukemia inhibitory factor is essential for subventricular zone neural stem cell and progenitor homeostasis as revealed by a novel flow cytometric analysis. Dev Neurosci 2012, 34:449–462. |
| [124] | Svendsen CN, Fawcett JW, Bentlage C, Dunnett SB. Increased survival of rat EGF-generated CNS precursor cells using B27 supplemented medium. Exp Brain Res 1995, 102:407–414. |
| [125] | Babu H, Cheung G, Kettenmann H, Palmer TD, Kempermann G. Enriched monolayer precursor cell cultures from micro-dissected adult mouse dentate gyrus yield functional granule cell-like neurons. PLoS One 2007, 2:e388. |
| [126] | Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15:501–513. |
| [127] | Vescovi AL, Reynolds BA, Fraser DD, Weiss S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron 1993, 11:951–966. |
| [128] | Shihabuddin LS, Ray J, Gage FH. FGF-2 is sufficient to isolate progenitors found in the adult mammalian spinal cord. Exp Neurol 1997, 148:577–586. |
| [129] | Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 1999, 208:166–188. |
| [130] | Podergajs N, Brekka N, Radlwimmer B, Herold-Mende C, Talasila KM, Tiemann K, Rajcevic U, Lah TT, Bjerkvig R, et al. Expansive growth of two glioblastoma stem-like cell lines is mediated by bFGF and not by EGF. Radiol Oncol 2013, 47:330–337. |
| [131] | Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL, Forsyth P, Auer RN, Dunn JF, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells 2009, 27:1722–1733. |
| [132] | Zhu L-L, Wu L-Y, Yew DT, Fan M. Effects of hypoxia on the proliferation and differentiation of NSCs. Mol Neurobiol 2005, 31:231–242. |
| [133] | Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci 2000, 20:7377–7383. |
| [134] | Mongiardi MP. Angiogenesis and hypoxia in glioblastoma: a focus on cancer stem cells. CNS Neurol Disord Drug Targets 2012, 11:878–883. |
| [135] | Ljungkvist ASE, Bussink J, Kaanders JHAM, van der Kogel AJ. Dynamics of tumor hypoxia measured with bioreductive hypoxic cell markers. Radiat Res 2007, 167:127–45. |
| [136] | Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J 2004, 381:761–767. |
| [137] | Bruick RK. Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev 2003, 17:2614–2623. |
| [138] | Berra E, Ginouvès A, Pouysségur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep 2006, 7:41–45. |
| [139] | Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 2006, 70:1469–1480. |
| [140] | Kolenda J, Jensen SS, Aaberg-Jessen C, Christensen K, Andersen C, Brünner N, Kristensen BW. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J Neurooncol 2011, 103:43–58. |
| [141] | Persano L, Pistollato F, Rampazzo E, Della Puppa A, Abbadi S, Frasson C, Volpin F, Indraccolo S, Persano L, et al. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1α stability and MGMT expression. Cell Death Dis 2012, 3:e412. |
| [142] | Panchision DM. The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol 2009, 220:562–568. |
| [143] | Méndez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang SC, Newcomb EW, Zagzag D. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol Cancer 2010, 9:133. |
| [144] | Koh MY, Lemos R, Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res 2011, 71:4015–4027. |
| [145] | Koh M, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci 2012, 37:364–372. |
| [146] | Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8:3274–3284. |
| [147] | Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev 2006, 20:557–570. |
| [148] | Platet N, Liu SY, Atifi ME, Oliver L, Vallette FM, Berger F, Wion D. Influence of oxygen tension on CD133 phenotype in human glioma cell cultures. Cancer Lett 2007, 258:286–290. |
| [149] | Mak AB, Blakely KM, Williams RA, Penttilä PA, Shukalyuk AI, Osman KT, Kasimer D, Ketela T, Moffat J. CD133 protein N-glycosylation processing contributes to cell surface recognition of the primitive cell marker AC133 epitope. J Biol Chem 2011, 286:41046–41056. |
| [150] | Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28:3949–3959. |
| [151] | McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133 +human glioblastoma cells in vitro. Mol Cancer Res 2009, 7:489–497. |
| [152] | Schiffer D, Mellai M, Annovazzi L, Caldera V, Piazzi A, Denysenko T, Melcarne A. Stem cell niches in glioblastoma: a neuropathological view. Biomed Res Int 2014, 2014:725921. |
| [153] | Gómez-Gaviro MV, Lovell-Badge R, Fernández-Avilés F, Lara-Pezzi E. The vascular stem cell niche. J Cardiovasc Transl Res 2012, 5:618–630. |
| [154] | Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 1995, 92:5510–5514. |
| [155] | Wang R, Jin F, Zhong H. A novel experimental hypoxia chamber for cell culture. Am J Cancer Res 2014, 4:53–60. |
| [156] | Kieninger J, Aravindalochanan K, Sandvik JA, Pettersen EO, Urban GA. Pericellular oxygen monitoring with integrated sensor chips for reproducible cell culture experiments. Cell Prolif 2014, 47:180–188. |
| [157] | Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol 2012, 181:1126–1141. |
| [158] | Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468:824–828. |
| [159] | Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468:829–833. |
| [160] | Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol 2005, 7:134–153. |
| [161] | Caspani EM, Crossley PH, Redondo-Garcia C, Martinez S. Glioblastoma: A Pathogenic Crosstalk between Tumor Cells and Pericytes. PLoS One 2014, 9:e101402. |
| [162] | Brat DJ, Van Meir EG. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest 2004, 84:397–405. |
| [163] | Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringnér M, Mörgelin M, Bourseau-Guilmain E, Bengzon J, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A 2013, 110:7312–7317. |
| [164] | Yan GN, Yang L, Lv YF, Shi Y, Shen LL, Yao XH, Guo QN, Zhang P, Cui YH, et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J Pathol 2014, 234:11–22. |
| [165] | Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, He X, Hervey-Jumper SL, Heth JA, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res 2011, 71:6061–6072. |
| [166] | Gürsel DB, Berry N, Boockvar JA. The contribution of Notch signaling to glioblastoma via activation of cancer stem cell self-renewal: the role of the endothelial network. Neurosurgery 2012, 70:N19–21. |
| [167] | Galan-Moya EM, Treps L, Oliver L, Chneiweiss H, Vallette FM, Bidère N, Gavard J. Endothelial secreted factors suppress mitogen deprivation-induced autophagy and apoptosis in glioblastoma stem-like cells. PLoS One 2014, 9:e93505. |
| [168] | Aihara M, Sugawara K, Torii S, Hosaka M, Kurihara H, Saito N, Takeuchi T. Angiogenic endothelium-specific nestin expression is enhanced by the first intron of the nestin gene. Lab Invest 2004, 84:1581–1592. |
| [169] | Infanger DW, Cho Y, Lopez BS, Mohanan S, Liu SC, Gursel D, Boockvar JA, Fischbach C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res 2013, 73:7079–7089. |
| [170] | Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508:55–60. |
| [171] | Dore-Duffy P, Katychev A, Wang X, Van Buren E. CNS microvascular pericytes exhibit multipotential stem cell activity. J Cereb Blood Flow Metab 2006, 26:613–624. |
| [172] | Montiel-Eulefi E, Nery AA, Rodrigues LC, Sánchez R, Romero F, Ulrich H. Neural differentiation of rat aorta pericyte cells. Cytom A 2012, 81:65–71. |
| [173] | Ochs K, Sahm F, Opitz CA, Lanz TV, Oezen I, Couraud PO, von Deimling A, Wick W, Platten M. Immature mesenchymal stem cell-like pericytes as mediators of immunosuppression in human malignant glioma. J Neuroimmunol 2013, 265:106–116. |
| [174] | Liu AY, Ouyang G. Tumor angiogenesis: a new source of pericytes. Curr Biol 2013, 23:R565–R568. |
| [175] | Scully S, Francescone R, Faibish M, Bentley B, Taylor SL, Oh D, Schapiro R, Moral L, Yan W, et al. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J Neurosci 2012, 32:12950–12960. |
| [176] | Simonavicius N, Robertson D, Bax DA, Jones C, Huijbers IJ, Isacke CM. Endosialin (CD248) is a marker of tumor-associated pericytes in high-grade glioma. Mod Pathol 2008, 21:308–315. |
| [177] | Dorshkind K, Pollack SB, Bosma MJ, Phillips RA. Natural killer (NK) cells are present in mice with severe combined immunodeficiency (scid). J Immunol 1985, 134:3798–3801. |
| [178] | Wiltrout RH, Brunda MJ, Holden HT. Variation in selectivity of tumor cell cytolysis by murine macrophages, macrophage-like cell lines and NK cells. Int J Cancer 1982, 30:335–342. |
| [179] | Yoshino H, Ueda T, Kawahata M, et al. Natural killer cell depletion by anti-asialo GM1 antiserum treatment enhances human hematopoietic stem cell engraftment in NOD/Shi-scid mice. Bone Marrow Transplant 2000, 26:1211–1216. |
| [180] | Ito M, Hiramatsu H, Kobayashi K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002, 100:3175–3182. |
| [181] | Donoghue JF, Bogler O, Johns TG. A simple guide screw method for intracranial xenograft studies in mice. J Vis Exp 2011, 26. pii:3157. |
| [182] | Lal S, Lacroix M, Tofilon P, et al. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg 2000, 92:326–33. |
| [183] | Patru C, Romao L, Varlet P, et al. CD133, CD15/SSEA-1, CD34 or side populations do not resume tumor-initiating properties of long-term cultured cancer stem cells from human malignant glio-neuronal tumors. BMC Cancer 2010, 10:1–11. |
| [184] | Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene 2004, 23:7267–7273. |
| [185] | Marshall GP, Reynolds BA, Laywell ED. Using the neurosphere assay to quantify neural stem cells in vivo. Curr Pharm Biotechnol 2007, 8:141–145. |
| [186] | Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8:486–498. |
| [187] | Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron 1994, 13:1071–1082. |
| [188] | Hu Y, Smyth G. ELDA: Limiting Dilution Analysis for stem cell research. Journal of Immunological Methods 2009. http://bioinf.wehi.edu.au/software/elda/. |
| [189] | Shin SI, Freedman VH, Risser R, Pollack R. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci U S A 1975, 72:4435–4439. |
| [190] | Neugut AI, Weinstein IB. The use of agarose in the determination of anchorage-independent growth. In vitro 1979, 15:351–355. |
| [191] | Mendiburu-Eliçabe M, Gil-Ranedo J, Izquierdo M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin Transl Oncol 2014, 16:495–502. |
| [192] | Gil-Ranedo J, Mendiburu-Eliçabe M, García-Villanueva M, Medina D, del álamo M, Izquierdo M. An off-target nucleostemin RNAi inhibits growth in human glioblastoma-derived cancer stem cells. PLoS One 2011, 6:e28753. |
| [193] | Zhen Y, Zhao S, Li Q, Li Y, Kawamoto K. Arsenic trioxide-mediated Notch pathway inhibition depletes the cancer stem-like cell population in gliomas. Cancer Lett 2010, 292:64–72. |
| [194] | Warrier S, Pavanram P, Raina D, Arvind M. Study of chemoresistant CD133+ cancer stem cells from human glioblastoma cell line U138MG using multiple assays. Cell Biol Int 2012, 36:1137–1143. |
| [195] | Ma YH, Mentlein R, Knerlich F, Kruse ML, Mehdorn HM, Held-Feindt J. Expression of stem cell markers in human astrocytomas of different WHO grades. J Neurooncol 2008, 86:31–45. |
| [196] | Pan GJ, Chang ZY, Schöler HR, Pei D. Stem cell pluripotency and transcription factor Oct4. Cell Res 2002, 12:321–329. |
| [197] | Kobayashi K, Takahashi H, Inoue A, Harada H, Toshimori S, Kobayashi Y, Goto K, Sugimoto K, Yano H, Ohnishi T, Tanaka J. Oct-3/4 promotes migration and invasion of glioblastoma cells. J Cell Biochem 2012, 113:508–517. |
| [198] | Jin X, Jin X, Jung J-E, Beck S, Kim H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem Biophys Res Commun 2013, 433:496–501. |
| [199] | Park D, Xiang AP, Mao FF, Zhang L, Di CG, Liu XM, Shao Y, Ma BF, Lee JH, et al. Nestin is required for the proper self-renewal of neural stem cells. Stem Cells 2010, 28:2162–2171. |
| [200] | Kitai R, Horita R, Sato K, Yoshida K, Arishima H, Higashino Y, Hashimoto N, Takeuchi H, Kubota T, et al. Nestin expression in astrocytic tumors delineates tumor infiltration. Brain Tumor Pathol 2010, 27:17–21. |
| [201] | Kukekov VG, Laywell ED, Thomas LB, Steindler DA. A nestin-negative precursor cell from the adult mouse brain gives rise to neurons and glia. Glia 1997, 21:399–407. |
| [202] | Griguer CE, Oliva CR, Gobin E, Marcorelles P, Benos DJ, Lancaster JR Jr, Gillespie GY. CD133 is a marker of bioenergetic stress in human glioma. PLoS One 2008, 3:e3655. |
| [203] | Beier D, Röhrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, Proescholdt M, Brawanski A, Bogdahn U, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res 2008, 68:5706–5715. |
| [204] | Fouse SD, Nakamura JL, James CD, Chang S, Costello JF. Response of primary glioblastoma cells to therapy is patient specific and independent of cancer stem cell phenotype. Neuro Oncol 2014, 16:361–371. |
| [205] | Johannessen T-CA, Bjerkvig R, Tysnes BB. DNA repair and cancer stem-like cells–potential partners in glioma drug resistance? Cancer Treat Rev 2008, 34:558–567. |
| [206] | Günther W, Pawlak E, Damasceno R, Arnold H, Terzis AJ. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br J Cancer 2003, 88:463–469. |
| [207] | Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, Lesniak MS, Ahmed AU. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ 2014, 21:1119–1131. |
| [208] | Riganti C, Salaroglio IC, Caldera V, Campia I, Kopecka J, Mellai M, Annovazzi L, Bosia A, Ghigo D, et al. Temozolomide downregulates P-glycoprotein expression in glioblastoma stem cells by interfering with the Wnt3a/glycogen synthase-3 kinase/β-catenin pathway. Neuro Oncol 2013, 15:1502–1517. |
| [209] | Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cell - much more complex than expected. Mol Cancer 2011, 10:128. |
| [210] | Blough MD, Westgate MR, Beauchamp D, Kelly JJ, Stechishin O, Ramirez AL, Weiss S, Cairncross JG. Sensitivity to temozolomide in brain tumor initiating cells. Neuro Oncol 2010, 12:756–760. |
| [211] | Ohba S, Hirose Y, Kawase T, Sano H. Inhibition of c-Jun N-terminal kinase enhances temozolomide-induced cytotoxicity in human glioma cells. J Neurooncol 2009, 95:307–316. |
| [212] | Okada M, Sato A, Shibuya K, Watanabe E, Seino S, Suzuki S, Seino M, Narita Y, Shibui S, et al. JNK contributes to temozolomide resistance of stem-like glioblastoma cells via regulation of MGMT expression. Int J Oncol 2014, 44:591–599. |
| [213] | Sato A, Sunayama J, Matsuda K, Seino S, Suzuki K, Watanabe E, Tachibana K, Tomiyama A, Kayama T, et al. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011, 29:1942–1951. |
| [214] | Johannessen TC, Prestegarden L, Grudic A, Hegi ME, Tysnes BB, Bjerkvig R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro Oncol 2013, 15:269–278. |
| [215] | Shi L, Wan Y, Sun G, Zhang S, Wang Z, Zeng Y. miR-125b inhibitor may enhance the invasion-prevention activity of temozolomide in glioblastoma stem cells by targeting PIAS3. Bio Drugs 2014, 28:41–54. |
| [216] | Chen J, Fu X, Wan Y, Wang Z, Jiang D, Shi L. miR-125b inhibitor enhance the chemosensitivity of glioblastoma stem cells to temozolomide by targeting Bak1. Tumour Biol 2014, 35:6293–6302. |
| [217] | Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9:391–403. |
| [218] | He J, Liu Y, Zhu TS, Xie X, Costello MA, Talsma CE, Flack CG, Crowley JG, Dimeco F, et al. Glycoproteomic analysis of glioblastoma stem cell differentiation. J Proteome Res 2011, 10:330–338. |
| [219] | Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444:761–765. |