| Journal of Cancer Stem Cell Research (2014), 2:e1004 © 2013 Creative Commons. All rights reserved ISSN 2329-5872 DOI: 10.14343/JCSCR.2014.2e1004 http://cancerstemcellsresearch.com |
|
| Journal of Cancer Stem Cell Research (2014), 2:e1004 © 2013 Creative Commons. All rights reserved ISSN 2329-5872 DOI: 10.14343/JCSCR.2014.2e1004 http://cancerstemcellsresearch.com |
|
| Review Article | Open Access |
| A concise review on the current understanding of pancreatic cancer stem cells | |
| Arokia Priyanka Vaz1, Moorthy P. Ponnusamy1, Parthasarathy Seshacharyulu1, and Surinder K. Batra1,2,* | |
| 1Department of Biochemistry and Molecular Biology, 2Eppley Institute for Research in Cancer and Allied Diseases and Buffett Cancer Center, University of Nebraska Medical Center, Omaha, NE, USA | |
| *Correspondence: Surinder K. Batra, Ph.D., Department of Bio-chemistry and Molecular Biology, Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, Nebraska, 68198-5870, USA. Phone: 402-559-5455, Fax: 402-559-6650; E-mail: sbatra@unmc.edu Received: July 5, 2014 Accepted: July 6, 2014 | |
Abstract: Several evidences suggest that a small population of cells known as cancer stem cells (CSCs) or tumor initiating stem-like cells within a tumor is capable of tumor initiation, maintenance and propagation. Recent publications have supported the existence of CSCs in pancreatic tumors. The pancreatic stem/progenitor cells, which express self-renewal markers, are identified to be present in the peribiliary gland. Based on the CSC hypothesis, mutations can lead to the transformation of stem/progenitor cells or differentiated cells into CSCs. The pancreatic CSCs express a wide array of markers such as CD44, CD24, ESA, CD133, c-MET, CXCR4, PD2/Paf1 and ALDH1. The CSCs are isolated based on surface markers or by other methods such as ALDEFLOUR assay or Hoechst 33342 dye exclusion assay. The isolated cells are further characterized by in vitro and in vivo tumorigenic assays. The most important characteristics of CSCs are its ability to self-renew and impart drug resistance towards chemotherapy. Moreover, these distinct cells display alteration of signaling pathways pertaining to CSCs such as Notch, Wnt and Shh to maintain the self-renewal process. Failure of cancer treatment could be attributed to the therapy resistance exhibited by the CSCs. Metastasis and drug resistance in pancreatic cancer is associated with epithelial to mesenchymal transition (EMT). Furthermore, mucins, the high molecular weight proteins are found to be associated with pancreatic CSCs and EMT. Understanding the underlying molecular pathways that aid in the metastatic and drug resistant nature of these distinct cells will aid in targeting these cells. Overall, this review focuses on the various aspects of pancreatic adult/stem progenitors, CSC hypothesis, its markers, pathways, niche, EMT and novel therapeutic drugs used for the elimination of pancreatic CSCs.
Keywords: Cancer stem cells, pancreatic cancer, niche, markers, signaling pathways, drug resistance.
ABCB1 - ATP-binding cassette, sub-family B (MDR/TAP), member 1)
CXCR4 - Cysteine-x-cysteine chemokine receptor 4
DCLK1 - Doublecortin-like kinase 1
SOX2 - Sex-determining region Y (SRY)-Box2
PDAC - Pancreatic ductal adenocarcinoma
CSC - Cancer stem cell
SP - Side population
NSP - Non side population
EMT - Epithelial to mesechymal transition
Pancreatic cancer is one of the most lethal cancers among all solid malignancies. According to the National cancer institute, it has been estimated that approximately 46,420 new cases and 39,590 deaths would be reported in the year 2014 [1]. The incidence rates have been increasing for pancreatic cancer over the past several years. Currently, pancreatic cancer has been listed as the fourth leading cause of death due to cancer and by 2020 it is predicted to be ranked as the second leading cause of cancer related deaths [2]. On the positive side, the survival rate has increased from 3% to 6.7% in the past 35 years. There are several risk factors associated with this disease. Primarily, cigarette smoking has been the largest known risk factor for pancreatic cancer development [3, 4]. Other well-known risk factors such as obesity, pancreatitis, diabetes and other forms of tobacco usage are associated with the development of pancreatic cancer. In addition, those individuals who have a strong family history of pancreatic cancer are more prone to an increased risk of developing pancreatic cancer [5]. Approximately, 5–10% of pancreatic ductal adenocarcinoma (PDAC) cases are hereditary with nearly 80% penetrance [6, 7]. Pancreatic cancer is not just a single entity caused by a single mutation; it has various precursors which arise due to multiple mutations.
The three precursors for pancreatic cancer are the highly occurring precursor; such as the pancreatic intraepithelial neoplasia (PanINs), and less commonly occurring precursors such as; intraductal papillary mucinous neoplasm (IPMN) and mucinous cystic neoplasm (MCN) [8]. Histologically, normal pancreas undergoes a series of morphological changes giving rise to low grade PanINs which eventually gives rise to high grade PanINs [9]. These PanIN lesions eventually develop into infiltrative adenocarcinoma [10]. Many genetic alterations were defined in pancreatic cancer such as earlier events including K-ras point mutation, EGFR overexpression and gene amplification and HER2/neu overexpression and later events such as inactivation of p16, p53, DPC4 and BRCA. Considering the genetic alterations, currently there are several animal models developed to study the progression of pancreatic cancer [9]. Animal models are developed to recapitulate the genetic alterations of the human pancreatic cancer and also they serve as a tool to understand the mechanisms underlying the disease.
In the recent past various animal models have been developed using the Cre-Lox technology such as Pdx1-Cre; LSL-KrasG12D, Ptf1/p48-Cre; LSL-KrasG12D and LSL-KrasG12D/+/Mist1Cre-ER/ [11–13]. Eventually, animal models harboring additional modifications such as inactivation/mutation of p16, p19, p53, transforming growth factor (TGFβ) and smad4 were developed [14–16]. These in vivo models help in understanding the progression of pancreatic cancer from lower to higher grade lesions which slowly develops to invasive carcinoma and finally to metastasis. Although several aspects of PDAC have been studied so far, the evidences for the emergence of pancreatic cancer from cancer stem cells have been quite limited but intriguing as well.
Cancer stem cells (CSCs) or tumor initiating stem-like cells (TICs) are a small subset of cancer cells which are capable of self-renewal and resist various chemotherapeutic drugs [17]. This sub-population behaves like stem cells by undergoing either asymmetric or symmetric cell division thereby maintaining its population within the cancer. CSCs have been identified in various cancers including brain, breast, ovarian, prostate, pancreatic and colon [18–25]. Simeone et al. [20], demonstrated the presence of CSCs in pancreatic cancer for the first time. Pancreatic CSCs were characterized by CD44+ CD24+ and ESA+ markers. Eventually, several pieces of evidence have cropped up to prove the existence of pancreatic CSCs [26–28]. These pieces of evidence emphasize the importance of identifying pancreatic cancer stem cells. Simultaneously, targeting these CSCs in pancreatic cancer has become another challenging area of interest. In this review article, we will summarize the earlier findings of pancreatic cancer stem cells, the potential techniques used to enrich and characterize pancreatic CSCs, pancreatic CSC niche, the various signaling pathways involved in the maintenance of pancreatic CSCs, drug resistance and EMT, mucins in pancreatic CSCs and the current strategies used to target pancreatic CSCs.
By the year 2006, many studies reported the existence of CSCs in various cancers [18, 22, 29]. After several years of CSC discovery, the first evidence for the existence of pancreatic CSCs was reported by two groups in the year 2007 [20, 30]. Li et al. [20], demonstrated that the CD44+CD24+ESA+ cells isolated from human PDAC could self-renew, had differentiation potential, and had enhanced Shh expression. Subcutaneous injection of 500 cells (positive for CD44, CD24 and ESA) in mice could generate tumors (7/12 mice) whereas implantation of pancreatic cancer cells negative for these markers could not. Equally significant, a second study showed the presence of pancreatic CSCs having the ability to metastasize. Notably, the CD133+CXCR4+ CSC subpopulation isolated from pancreatic tumors displayed metastatic activity [30]. Emerging evidence demonstrates that the ZEB1-microRNA200 feedback loop is essential to promote the migratory CSCs in pancreatic cancer [31].
Later in 2011, c-Met was identified as an important CSC marker in pancreatic cancer [28]. Strikingly, the c-Met expressing CSCs (c-Methigh) had the ability to give rise to a larger tumor as opposed to no tumor formation in the c-Met negative cells. A c-met inhibitor such as XL184 could reduce the CSC population [28]. Subsequently, Van den Broeck et al. [26], used a different method to study the pancreatic CSCs [26]. They have isolated side population (SP) and non-side population (NSP) from PDAC surgical resection specimens using the Hoechst 33342 dye based FACS analysis. Two important genes such as ABCB1, a multidrug resistance transporter as well as CXCR4, a chemokine receptor were found to be upregulated in the SP fraction as opposed to the NSP fraction. They also demonstrated that these two genes have been associated with the worst patient survival. It has been suggested that this subpopulation of cancer cells such as the CSCs should be the prime target for therapy.
A recent study demonstrated that SOX2, a transcription factor which plays a role in the embryonic development has been found to cause de-differentiation thereby imparting stem cell-like characteristics to pancreatic cancer cells. SOX2 is absent in the normal acinar or ductal compartment. However, its expression has been observed in 19.3% of human pancreatic tumors. The study suggested that SOX2 positive cancer cells could serve as an essential therapeutic target; as its expression has significantly increased in the ESA+/CD44+ CSC population, and is also found to regulate genes controlling EMT and G1/S transition thereby contributing to dedifferentiation and stemness [32]. The latest work by Bailey et al. [27], demonstrated the existence of a distinct population of pancreatic cancer initiating cells in KCPdx, KCiMist1 and KPCPdx mice expressing DCLK1 which is a microtubule regulator. They have also demonstrated that pancreatic CSCs could be identified at very early stages such as in PanIN 1 (Pancreatic intraepithelial neoplasia-1) in KPC mice. Altogether, these evidences clearly validate the presence of CSC subpopulation in pancreatic cancer.
During the embryonic developmental stage, pancreas develops as dorsal and ventral evaginations from the foregut endoderm in the 5th week of gestation [33]. Cells from the dorsal and ventral buds slowly undergo lineage commitment to either of the two compartments such as the endocrine and the exocrine compartments. The endocrine compartment comprises the islets while the exocrine compartment is organized into acinar, ductal and centroacinar cells (Figure 1) [34]. In addition to the above mentioned compartments, a novel gland like mucinous compartment known as the pancreatic ductal gland has been identified to possess a characteristic molecular signature [35]. With different compartments present in the pancreas, the question is from where do the pancreatic progenitors arise?
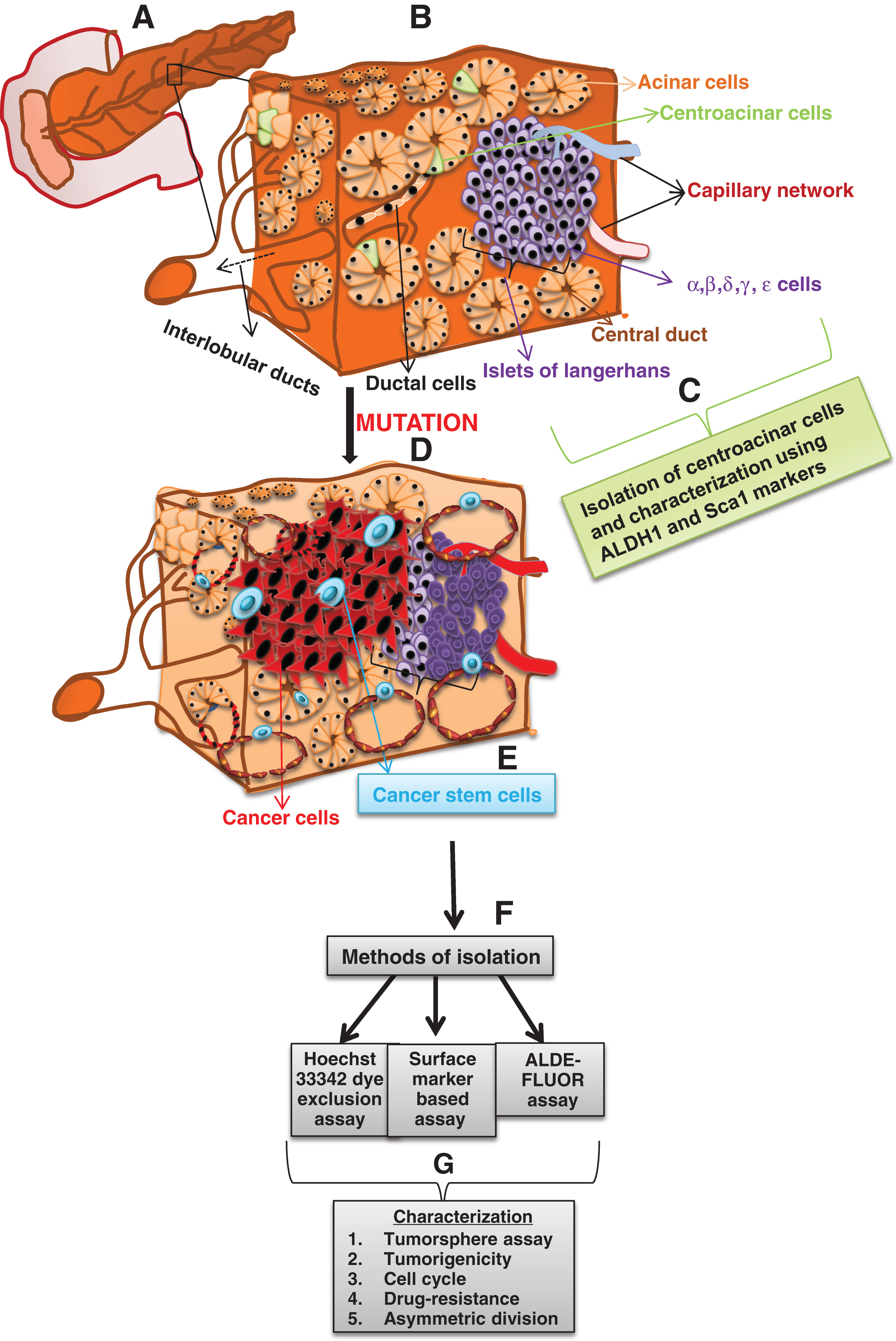 |
Figure 1. A comprehensive diagram depicting the evolution of cancer stem cells from normal pancreas due to accumulation of mutations, followed by its isolation and characterization. (A) A simplistic representation of the adult pancreas. (B) A cross section of the pancreas illustrates the important components of the pancreas such as the acini, ductal cells, centroacinar cells, islets of langerhans comprising the alpha, beta, delta, gamma and epsilon cells. (C) Isolation of the centro acinar cells using ALDH1 and Sca 1. (D) The normal cells undergo mutation which may give rise to cancer stem cells. (E) The net result of mutation in either the stem cell, progenitor cell or the differentiated cells leads to the formation of cancer stem cells. (F) Cancer stem cells are then isolated using various methods such as the hoechst 33342 dye exclusion assay, surface marker based isolation and the aldefluor assay. (G) The isolated cancer stem cells are characterized using various methods such as tumorsphere assay, tumorigenicity assay, cell cycle analysis, the ability to undergo asymmetric division and the ability to withstand drug pressure. |
Pancreas is an essential organ whose size is controlled by the size of the progenitor population that is present in the developing pancreatic bud [36]. On the other hand, the reduction in the number of progenitor cell population does not control the size of the liver during developmental stages [36]. Results showed the ability of pancreatic progenitors to grow, divide and differentiate after a reduction in the Pdx1 progenitor pool in mice. However, it could not increase the cell division rate in order to make a normal sized organ [36]. Progenitors isolated from mice are found to bear several surface markers. For instance, Samuelson et al. [37], showed that the highly proliferative pancreatic progenitor population isolated from mice has been found to express stem cells antigen 1- (Sca-1). Another study showed the presence of Nestin positive multipotent progenitor cells in the centrilobular ducts of the adult rat pancreas [38]. Interestingly Smukler et al., demonstrated the presence of insulin positive multipotent stem cells which had the ability to divide, thereby contributing to both pancreatic and neural lineages [39].
Recent reports propose that the biliary tree derived cells are the precursors of pancreatic committed progenitors [40]. There is evidence for the presence of pancreatic stem cells and/or progenitors in the peribiliary gland (PBG) which connects to the pancreatic duct glands within the pancreas [40]. The stem cells in the peribiliary gland are highly proliferative and they express pluripotency markers such as NANOG, OCT4, and SALL4 but do not express mature pancreatic markers [40].
Notably, Rovira et al. [41], demonstrated that ALDH1 expressing centroacinar cells behave like adult/stem progenitor cells. Evidences for the origin of CSCs in pancreas are very limited. A recent study demonstrated that the centroacinar cells; which is located at the junction of acini and ducts, has been suggested to be the origin of PanINs and pancreatic ductal adenocarcinoma and since these cells express stem cell markers it could be proposed that CSCs could arise from the centroacinar cells [42]. Recent evidence also demonstrated that DCLK1 expressing cells in the Kras; p53; PdxCre mouse tumors show CSC like phenotype which may have originated from centroacinar cells [27]. Apart from the centroacinar cells, the differentiated acinar cells could be an essential source of stem cells as these cells are found to harbor facultative progenitor characteristics. Under favorable circumstances such as organ injury these facultative progenitors attains a precursor phenotype [43]. In the future, many studies are required to prove the concept of CSCs origin from adult pancreatic stem/progenitor cells.
Stem cells survive in a niche which provides favorable conditions for it to self-renew. Similarly, a tumor is governed by its microenvironment/niche which encompasses several components such as the cancer associated fibroblasts, CSCs, immune cells, signaling molecules, blood vessels and the extracellular matrix. It has been identified that tumor stroma is composed of pancreatic stellate cells which undergoes the paracrine Nodal/Activin signaling thereby forming a paracrine niche for pancreatic CSCs. It was reported that the pancreatic stellate cells secrete the embryonic morphogens Nodal/Activin. These secretions were found to support the in vitro sphere formation and promote invasiveness of pancreatic CSCs [44]. Hamada et al. [45], has shown that the presence of stellate cells improved the spheroid forming ability of cancer cells and the expression of CSC related genes such as Nestin, ABCG2 and LIN28 was induced. Hence, the cross talk between the niche and the CSCs remains pivotal.
Due to the technical difficulties in isolating exclusively the CSC population, several methods have been employed to solely enrich the CSC population from the heterogeneous cancer cells. The methods used are aldefluor assay, Hoechst 33342 dye method and surface marker based isolation (Figure 1).
This assay has been developed based on the increased aldehyde dehydrogenase (ALDH) activity in hematopoietic stem cells. ALDH is required for the oxidation of intracellular aldehydes thereby resulting in the oxidation of retinol to retinoic acid [46]. The aldefluor assay employs an ALDH fluorescent substrate called BODIPY-aminoacetaldehye (BAAA). BAAA passively diffuses into the living cells and gets converted into BODIPY aminoacetate (BAA-) by the intracellular ALDH. BAA- is retained inside the cells until it is effluxed by ATP binding cassette (ABC) transporters [47]. To determine the background fluorescence an ALDH inhibitor, Diethylaminobenzaldehyde (DEAB) is used. Using this assay, Rasheed et al. [48], claimed that the ALDH+ cells have enhanced tumorigenic potential and they are comparatively more invasive than the CD44+CD24+ pancreatic CSCs. Likewise Kim et al. [49], reported that the ALDHhigh cells are highly tumorigenic compared to the CD133+ and ALDHlow cell population. Gemcitabine treated xenograft tumors showed an enrichment of ALDH1 positive cells suggesting that they can tolerate chemotherapy similar to CSCs [50].
This method is one of the most common methods employed to isolate the side population (SP); based on its dye efflux properties in various types of cancer cells. SP cells constitute a subpopulation of cancer cells that can efficiently efflux the fluorescent DNA binding dye, Hoechst 33342, by an ATP binding cassette (ABC) transporter. This assay was initially employed to isolate SP cells from rat C6 glioma cell line [51]. As the SP cells exhibit higher tumorigenicity than non-SP cells it is believed that this method is used to detect CSCs. As a control for sorting the CSCs, an ABC transporter inhibitor; such as verapamil or reserpine, is used in order to determine the SP gate. These DNA binding dyes inhibit the efflux of the Hoechst dye by SP cells thus serving as an essential control. The main limitation of using Hoechst dye is its toxicity to cells; however, if the concentration and incubation time has been standardized the level of toxicity could be minimized. Small differences in cell densities, dye concentrations and staining timings may affect the phenotype of the SP cells. Despite these limitations, some researchers prefer to use the SP method, or the marker independent method, as it overcomes the barrier of using diverse CSC markers for isolation. By using the hoechst 33342 dye exclusion assay reports clearly show the presence of SP and NSP in various cancers such as brain, lung, prostate and pancreatic [26, 52–54].
Pancreatic CSCs can be isolated from cell lines or primary tumors using the markers detailed below.
c-Met belongs to the receptor tyrosine kinase family and is expressed in both normal and cancer cells [55]. The ligand associated with this receptor is known as hepatocyte growth factor (HGF). It has been reported that pancreatic cancer cells expressing high levels of c-Met (c-Methigh) displayed increased self-renewal capacity and tumorigenic potential as opposed to the non-expressing or c-Metlow expressing cancer cells [28]. Inhibition of c-MET using either small hairpin RNA or c-Met inhibitor resulted in decreased tumor growth. This work introduces c-Met as an essential CSC marker in pancreatic cancer.
CD24 and CD44 are cell surface glycoproteins involved in cell-cell interactions and cell adhesion. Epithelial specific antigen (ESA) which is also known as EpCAM is a widely used marker for CSCs isolation in various cancers [56]. Using these three markers, Li et al. [20], has demonstrated the existence of pancreatic CSCs. They have isolated the CD44+/CD24+/ESA+ pancreatic CSCs from the pancreatic tumors which accounted for 0.2–0.8% of pancreatic cancer cells that displayed the CSC features such as the self-renewal property and enhanced tumorigenic potential as opposed to the marker-negative population.
CD133 also known as Prominin1/AC133 is a surface glycoprotein expressed in the progenitor cell populations and it is a marker of CSCs of various cancer origins. CD133 is found to be expressed in pancreatic CSCs as demonstrated by Hermann et al. [30]. They clearly showed that CD133+CXCR4+ CSCs were responsible for the metastatic phenotype of the tumor and on depletion of the CSCs carrying these signature markers; it resulted in the abrogation of metastatic nature of pancreatic tumors [30].
Apart from the aforementioned markers, ALDH1 is one of the widely used markers to isolate pancreatic CSCs. In addition, CXCR4+ was used to denote a subset of CD133+ pancreatic CSCs which was associated with metastasis as well as drug resistance. Recently, a novel marker pancreatic differentiation 2 (PD2) was identified to maintain the self-renewal and drug resistance properties of pancreatic CSCs [57]. One of the most recent studies explored a novel marker integrin αvβ3 as a CSC driver in lung, breast, and pancreatic cancers which are highly resistant to erlotinib; a tyrosine kinase inhibitor [58]. The Kras-RalB-NF-κB pathway and the expression of the integrin were identified to be important for the initiation of tumor, self-renewal, anchorage independence and the resistance developed against erlotinib. Altogether, these markers could solely enrich the CSC population from a heterogeneous cancer cell population (Figure 2). Isolated CSCs are subsequently characterized for its self-renewal and tumorigenic properties.
 |
Figure 2. A schematic representation of various pancreatic cancer stem cell markers. Pancreatic CSC markers expressed on the cell surface comprises markers such as CD133, CD44, CD24, CXCR4, c-MET, ESA and DCLK1 and intracellular markers such as ALDH1 and PD2. Multidrug transporters belonging to the ATP binding cassette (ABC) superfamily expressed on the pancreatic CSCs aids in effluxing the Hoechst 33342 dye during the CSC isolation, thus mirroring the mechanism through which the chemotherapeutic drugs are being effluxed by the CSCs in pancreatic cancer. |
Once the CSCs are isolated using any of the previously mentioned methods these cells are characterized using the following assays:
To demonstrate the self-renewal capacity and the tumorigenic potential of the CSCs an in vitro tumorsphere assay is performed. One way to demonstrate the clonogenicity of the CSCs or the SP fraction is by seeding them in few numbers in a low attachment plate (with appropriate replicates), which are further allowed to grow for approximately 2 weeks. The total number of spheres formed is counted and these primary spheres are then subjected to serial dilution in order to demonstrate its self-renewal property in the secondary generation. The true CSC population will have the ability to form spheres faster than the primary generation.
In order to assess the tumorigenic potential of the CSCs, these distinct cells are injected in NOD-SCID mice, nude mice or NSG mice. In various cancers it has been reported that any number between 1 to <1000 CSCs when injected in mice have the ability to form a tumor [22, 59]. In pancreatic cancer, it has been shown that 50% of the mice developed tumor when 100 CD44+CD24+ESA+ cells were injected in mice [20]. The primary tumors are digested with collagenase and trypsin and CSCs isolated from these tumors are injected into the secondary recipients [48]. These mice should have developed the tumors even faster than that of the primary generation. Therefore, these assays are essential to be carried out in order to prove that the isolated CSCs are a true population (Figure 1).
Mucins are heavily glycosylated proteins which form a protective barrier to the cell surface. They are characterized by a heavily O-glycosylated tandem repeat region, rich in proline (P), threonine (T) and serine (S) residues also known as the PTS domain. The slow transition from a healthy to diseased state in pancreatic cancer is accompanied by an altered expression and localization of mucins [60]. There are as many as 21 members in the mucin family which are mainly divided into transmembrane and gel forming proteins. So far, two of the transmembrane mucins such as MUC1 and MUC4 have been found to be associated with cancer stem cells [61, 62].
In pancreatic cancer, it has been demonstrated that the down-regulation of MUC4 results in sensitizing the pancreatic cancer stem/progenitor cells to chemotherapeutic drugs, thus serving as an important therapeutic means in pancreatic cancer treatment [62]. Followed by this finding it was identified that in ovarian cancer, MUC4 was overexpressed in ovarian cancer cell line SKOV3, which led to the increased expression of HER2. This in turn resulted in increased CD133+ population as well as side population [63]. In the recent past, MUC1 was identified to be a potential marker in pancreatic and breast cancer stem cells. Engelmann et al. [64], has identified that around 77% of breast CSCs isolated using the Hoechst 33342 dye method were found to be MUC1bright cells. Similarly in pancreatic cancer, Curry et al. [61], has identified that 80% of the CSCs in patient samples expressed MUC1. Two sets of CSC populations were isolated from pancreatic cancer cell lines such as BXPC3 and Panc-1 using the triple marker such as CD44+CD24+EpCAM+ and the CD133+ cells. CSCs isolated using the triple marker sorting were up to 46.7% and 19.8% in BXPC3 and Panc-1 cell lines respectively. MUC1 expression was found to be detected at higher levels in both the populations [61].
Mucins have gained significant importance in pancreatic cancer research. Therefore, it will be important to explore mucins with respect to CSCs in the near future. Apart from MUC1 and MUC4, other mucins such as MUC5AC, MUC16 and MUC17 are yet to be explored from the cancer stem cell viewpoint.
Since self-renewal is a common feature of normal stem cells and CSCs, it is reasonable to believe that these cells share the same signaling pathways. The following signaling pathways such as Notch, Shh and Wnt play an important role in the pancreatic CSCs.
In the normal pancreas, Notch signaling controls the balance between the self-renewal and differentiation processes [65]. Additionally, Notch signaling is important for the pathogenesis of human cancers including pancreatic. Studies showed that the overexpression of Notch-1 resulted in increased clonogenicity, migration, invasion and induction of EMT phenotype in Aspc-1; a pancreatic cancer cell line. Moreover, the overexpression of Notch-1 resulted in a significant increase in the pancreatosphere formation which concomitantly expressed higher levels of the CSC markers, EpCAM and CD44 [66]. Bao et al. [66], has identified that Notch-1 signaling is crucial for the acquisition of EMT phenotype. Likewise, Abel et al. [67], has identified that Notch pathway is essential for the maintenance of pancreatic CSC population. They have observed that knockdown of Hes1 using shRNA and inhibition of the Notch pathway components by gamma secretase resulted in the reduction of the self-renewal capacity of pancreatic CSCs. Altogether, these studies clearly suggest that Notch signaling is important for the pancreatic CSC formation (Figure 3).
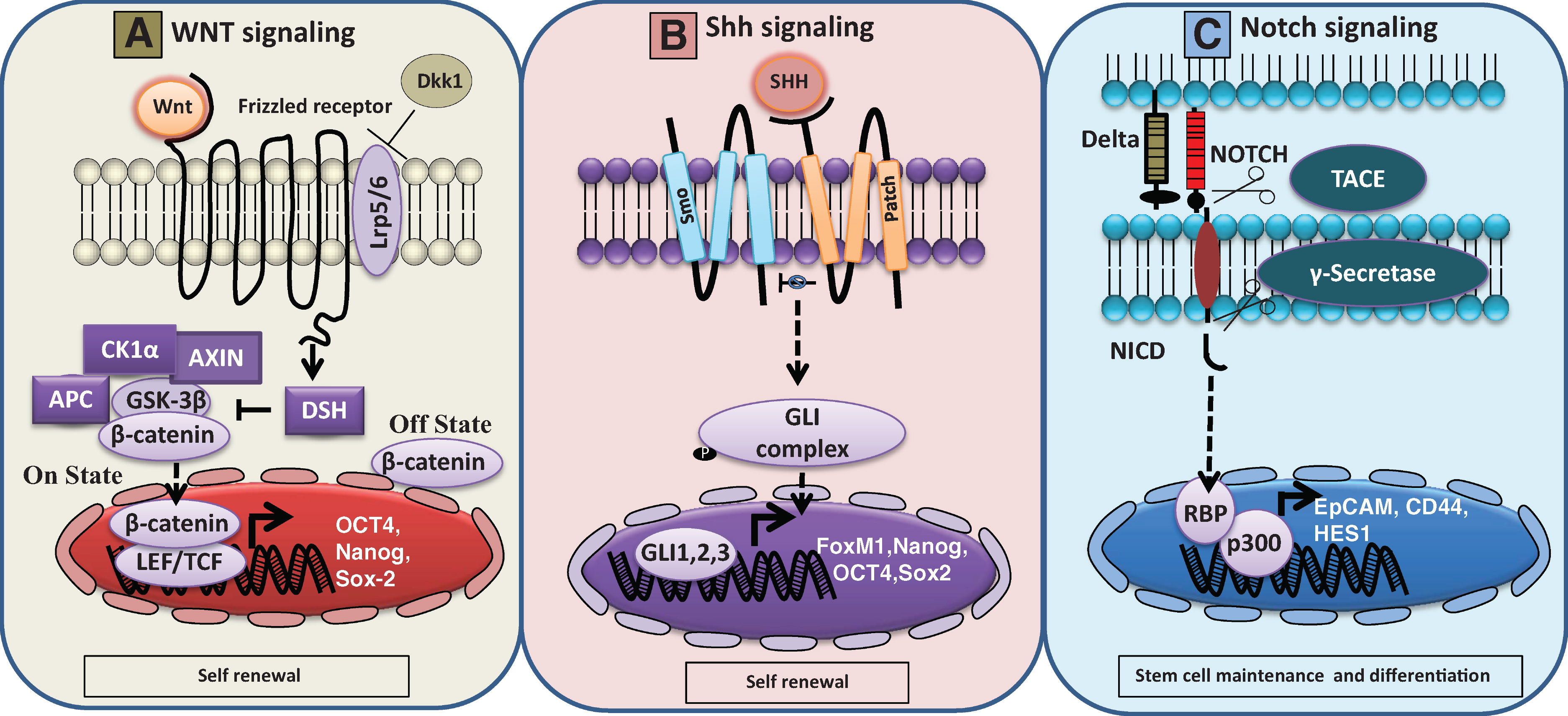 |
Figure 3. Schematic representation of Wnt, Shh and Notch signaling cascades in normal stem cells and pancreatic cancer stem cells. A. Wnt signaling pathway: Wnt proteins are secreted glycoproteins or ligands that transduces extracellular message to intracellular signaling cascade by binding through frizzled receptors. In the absence of this ligand (off state), β-catenin is sequestered by a complex of molecules such as Axin1, 2/APC/CK1α and GSK-3β, which is commonly known as destruction complex. Phosphorylation of β-catenin within this complex leads to ubiquitin and proteosomal mediated degradation process. In the presence of WNT ligand (on state), WNT protein binds to frizzled receptors along with its LRP5/6 co-receptor complex leading to activation of cytosolic phosphoprotein disheveled (DSH) resulting in interruption of the destruction complex. The activated DSH will inhibit GSK-3β activity which in turn leads to cytosolic accumulation of β-catenin, subsequently leading to nuclear translocation resulting in target genes activation. In normal stem cells, the Wnt pathway signaling causes activation of its target genes such as OCT4, Nanog and Sox-2 leading to maintenance of self-renewal property [89]. In addition, Wnt pathway is inhibited by Dickkopf 1((DKK1), a soluble Wnt inhibitor) resulting in reduction of stem cell population [90]. B. Shh signaling pathway: Sonic hedgehog (Shh) is a secreted and lipid modified ligand that binds to a transmembrane spanning receptor known as patched (Patch) leading to subsequent signal transduction events. In the absence of Shh ligand, Patch will constitutively repress smoothened (smo), another transmembrane spanning protein, having homology similar to G-protein coupled receptor (GPCR). Upon Shh ligand binding to Patch, smo inhibition by patch will be released, which subsequently leads to activation of downstream GLI (GLI1, 2, 3) family of transcription factors. In pancreatic cancer cells, GLI transcription factor activation leads to subsequent upregulation of Shh target genes such as FoxM1, Nanog, OCT4 and Sox2 [91–93]. These markers play a major role in the self-renewal nature of pancreatic cancer stem cells. C. Notch signaling pathway: In pancreatic cancer stem cells, notch signaling cascade plays a vital role in stem cell maintenance and differentiation process. Notch receptor is composed of an extracellular ligand binding domain, a single transmembrane spanning region and intracellular domain. Activation of notch signaling takes place through binding of delta ligand with notch receptor between neighboring cells. Upon ligand binding to notch receptor, it will undergo a conformational change that allows cleavage at extracellular portion of notch by a metalloprotease TNFα converting enzyme (TACE). Subsequently, the intracellular portion of notch will also be cleaved by Γ-secretase, an intramembrane protease thereby releasing notch intracellular domain containing portion (NICD). In pancreatic cancer cells, NICD will translocate into the nucleus and interacts with its transcription factor RBP and co activator p300 leading to activation of Epcam, CD44 and Hes1 genes [66, 67]. |
Hedgehog signaling pathway is essential for cell differentiation and tissue patterning events during the embryonic development of the pancreas [68]. Among the three hedgehog genes such as Sonic hedgehog (Shh), Indian hedgehog (Ihh) and Desert hedgehog homolog (Dhh), Shh shows the widest range of expression [68]. One of these three ligands binds to the receptor Patched1, which relieves the protein smoothened (Smo) from inhibition. Smo triggers the activation of the downstream target genes such as GLI family of transcription factors and PTCH (Figure 3). It has been reported that a nine fold increase in Shh mRNA levels has been found in the CD44+CD24+ESA+ cells when compared to the unsorted pancreatic cancer cells [20]. Sonic hedgehog- Gli signaling is identified to be essential for the pancreatic CSCs. Sulforane (SFN), an active component in cruciferous vegetables, was found to inhibit the self-renewal capacity of pancreatic CSCs by blocking the hedgehog pathway [69].
In addition to the above mentioned pathways, there is another pathway which is essential for the signaling in pancreatic CSCs. During embryonic development the Wnt-β-catenin signaling pathway plays an important role at different stages of pancreatic organogenesis. However, inhibition of this pathway is necessary for pancreatic specification during the early endoderm development [70]. Canonical Wnt signaling is found to be important for the progression of pancreatic cancer [71]. It has been reported that in colorectal cancer Wnt signaling is associated with EMT process and was found to activate a transcription factor snail thereby facilitating EMT. Snail is found to interact with β-catenin which is required for its activation. Since EMT is a process present in CSCs these findings suggest that β-catenin may have a role in pancreatic CSCs (Figure 3) [72]. However, in the future more studies are required to prove the role of β-catenin in pancreatic CSCs.
Apart from the three important signaling pathways there are other pathways which are involved in the maintenance of pancreatic CSCs. A recent study has reported that the inhibition of mTOR pathway by Rapamycin resulted in decreased viability of CD133+ pancreatic cancer cells and reduced the sphere forming ability of pancreatic cancer cells. These results suggest that the mTOR pathway is essential for the self-renewal of pancreatic CSCs [73]. Another study claims that the NF-κB pathway is highly activated in pancreatic CSCs. It was shown that treatment with NF-κB pathway inhibitors interrupts the stem cell-like properties [74]. Altogether, several signaling pathways have been identified to play significant roles in conserving the cancer stem cell phenotype in pancreatic cancer.
The most important property of CSCs is to acquire the EMT induced stemness phenotype which then leads to drug resistance to various chemotherapeutic agents. It has been well evidenced that human pancreatic cancer consists of a subset of cells; known as the side population, which is highly resistant to gemcitabine, a very commonly used chemotherapeutic agent in pancreatic cancer therapy [75]. This minor subset of cells displayed an increased expression of genes associated with epithelial-mesenchymal transition (SNAI2, LEF1), apoptotic regulation (FASLG, ETS1) and multi-drug resistance (ABCG2 and ABCA9) [75]. The cancer cells become resistant to drugs partly due to the acquisition of EMT phenotype [17]. It has been identified that the sensitivity of cancer cells is attributed by the EMT process. The epithelial marker such as E-cadherin was found to be strongly expressed in the gemcitabine sensitive pancreatic cancer cells whereas the gemcitabine resistant cells expressed mesenchymal markers such as vimentin and Zeb-1 [76]. Zeb1, a transcriptional suppressor has been identified to be an important player in the process of EMT. On silencing Zeb-1 in the mesenchymal cell lines, the expression of the epithelial markers such as E-cadherin, EVA1 and MAL2 was increased and most importantly the pancreatic cancer cells gained sensitivity to chemotherapeutic drugs [77]. Another report showed that pancreatic cancer cell lines; such as AsPC-1, MIAPaCa-2, PANC-1, Hs766T and MPanc96 cells which were resistant to three different chemotherapeutic drugs (gemcitabine, cisplatin and 5-fluorouracil), displayed EMT phenotype [77]. The above mentioned reports strongly suggest that drug resistance is associated with EMT phenotype. The migrating cancer progenitor cells play an important role in cancer progression and metastasis [78]. Likewise, another study showed that the gemcitabine resistant pancreatic cancer cells which display EMT characteristics showed down-regulation of single stranded small non coding RNAs namely micro RNAs (miRNA) including miR-200b, miR-200c, let-7(b-e) when compared to the gemcitabine sensitive pancreatic cancer cells. On re-expression of miR-200 in gemcitabine resistant pancreatic cancer cells, EMT markers such as ZEB1, vimentin and slug were down-regulated [76]. This suggests that miRNAs are important regulators in determining the EMT phenotype. Another study showed that miRNAs such as miR99a, miR100, miR-125b, miR-192 and miR-429 were differentially expressed in pancreatic CSCs. These miRNA clusters were found to be associated with the stem cell associated mRNAs in pancreatic CSCs [79]. Overall, these studies suggest that drug resistance and EMT are inter related and they play an important role in the maintenance of CSCs in pancreatic cancer.
Pancreatic cancer remains to be one of the most challenging cancers due to its intrinsic and extrinsic drug resistance, thereby leading to invasive carcinoma. Novel drugs are being synthesized to combat this disease. CSCs are a challenging factor for the chemotherapeutic treatments including pancreatic cancer. Metformin is one of the most significant drugs reported to have decreased the CSC population as evidenced by the diminished expression of CSC markers such as CD133, CD44, CXCR4 and SSEA-1 and self-renewal associated genes such as Nanog, Oct-4 and Sox2 [80]. Metformin was able to increase the reactive oxygen species production in CSCs and reduce its mitochondrial transmembrane potential. The in vitro tumorsphere assay revealed a significant decrease in the size and number of metformin treated spheres. Interestingly, they have shown that metformin retarded the formation of secondary and tertiary tumorspheres by hampering the self-renewal capacity of these CSCs. In cancer cells, the mode of action of this drug is by indirect activation of AMP-activated protein kinase (AMPK) signaling followed by inhibition of the mTOR activity thereby resulting in reduced cell proliferation and protein synthesis whereas an AMPK/mTOR independent pathway occurs in CSCs [80].
Another important drug named Salinomycin has been extensively used in the field of CSCs. Salinomycin is identified to target CD133+ pancreatic CSCs. A combinatorial effect of Salinomycin and gemcitabine has been used to eradicate pancreatic cancer in xenograft mice [81]. The combination of both drugs had an improved effect against CSCs over the individual agents itself. This suggests that administration of Salinomycin could therapeutically improve the efficacy of gemcitabine for the treatment of pancreatic cancer [81].
Sorafenib (SO), a multikinase inhibitor was used for targeting pancreatic CSCs. Studies demonstrated that SO administration led to the decreased spheroid formation, clonogenicity, ALDH1 activity, proliferation, angiogenesis and induced apoptosis. On the other hand, it also led to increased survival and regrowth of spheroid due to the SO induced activation of NF-kB. Therefore, in addition to SO, Sulforaphane (SF); a broccoli isothiocyanate, was also used to efficiently target pancreatic CSCs. This combinatorial treatment efficiently abolished SO-induced NF-kB binding which in turn led to abrogated spheroid formation, ALDH1 activity, clonogenicity, induction of apoptosis and tumor size reduction [82].
A novel drug namely cabozantinib (XL184) has been identified to inhibit c-MET, a recently established pancreatic CSC marker. Cabozantinib, a FDA approved drug decreased the viability and spheroid formation and also induced apoptosis in cancer cells. It also inhibits self-renewal property and the expression of CSC markers including SOX2, c-Met and CD133. Strikingly, cabozantinib increased the sensitivity of gemcitabine resistant cells. When this drug was administered to 330 medullary thyroid carcinoma patients, several side effects such as diarrhea, weight loss, loss of appetite, oral pain, nausea, hypertension, and hair color changes were reported. Regardless of these side-effects, this drug inhibited tumor progression and led to reduced tumor size in some patients [83].
Recent work by Zeng et al. [84], have demonstrated the synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 on pancreatic cancer and pancreatic CSCs. Together, these drugs target the chemoresistant cancer cells and CSCs. Therefore, novel drugs causing minimal side effects and maximal targeting of CSCs is the current need in the field of pancreatic CSCs. Moreover, there is a significant need in the area of developing small molecular inhibitors and nanoparticles targeted against CSCs to reduce the expression of the overexpressed proteins solely in CSCs. It is extremely important to design and improve the combinatorial therapies which could target the bulk of the tumor cells, CSCs and the residual dormant cells. It is well evident by now that CSC markers such as CD44, CD133 and CD24 are upregulated in pancreatic CSCs (Figure 2). Thus, raising antibody against CSC surface markers would be a major tool to target pancreatic CSCs. For example, antibody raised against CD44 led to the inhibition of pancreatic tumor initiation and postradiation recurrence in mice [85].
Wang et al. [86], reported that successful targeting of pancreatic CSCs could be achieved by targeting Notch using natural agents such as genistein, curcumin, quercetin and sulphorane. It has been identified that chloroquine targets pancreatic CSCs by inhibiting CXCR4 and hedgehog signaling [87]. Thus, based on the above mentioned reports it could be suggested that novel strategies encompassing combinatorial therapies could be used to achieve improved treatment outcome for pancreatic cancer patients.
The uncontrolled expansion of self-renewing CSCs results in cancer. Extensive studies over the past several years revealed the importance of this small subset of cells that could sustain the tumor. Although there are several methods employed to isolate CSCs, there are limitations with each of the currently used methods. Therefore, there is a need to identify improved methods for isolating purely the CSC population. Markers such as CD44, CD133 and ESA have been well established in pancreatic cancer but they serve as markers for other cancers as well. It is of utmost importance to identify specific markers which aid in the maintenance of pancreatic CSCs. As every organ has a specific gene expression pattern, it would be ideal to identify the specifics of pancreatic cancer. In the past, the identification of circulating tumor cells opened a new chapter in the field of cancer. The methods employed for the detection of tumor cells circulating in the blood stream are crucial. The most current methods used are based on the surface marker expression such as EpCAM. Similarly, if the CSCs have a sequence of signature markers expressed on its surface specific for each type of cancer, it enables the identification of CSCs, thereby facilitating easy targeting of these cells.
Given that very few CSCs when injected in mice can give rise to tumor much faster than the cancer cells, successful targeting of CSCs with a combination of chemotherapeutic agents could likely yield dramatic results. Besides CSCs, the players of the tumor microenvironment facilitate the pathogenesis of pancreatic cancer. As a result, it can be suggested that tumor microenvironment be considered as a crucial site for drug delivery. The most effective way of targeting pancreatic cancer is by destroying the CSC niche or by altering the expression of the important players which support the survival of CSCs. In the future, in vivo animal studies which explore the biology of pancreatic CSCs are required.
The signaling pathways such as Notch, Wnt and Shh are altered in CSCs. Therefore, clinical trials should focus on novel therapeutic agents that target CSCs and the important molecules in the signaling pathways in order to control the aggressiveness of pancreatic cancer. Reversal of EMT phenotype will aid in the treatment of pancreatic cancer. Different clonogenic CSCs have been identified in many other cancers in the recent past. It is also important to identify the clonogenicity of aggressive CSCs in pancreatic cancer. Origin of CSCs is one of the emerging fields; hence it is also important to identify the specific origin of pancreatic CSCs in order to target the CSCs.
The major problem with pancreatic cancer is tumor recurrence. Once the drug is withdrawn or due to the development of resistance towards drugs, the cancer reappears. Therefore, there is a need to elucidate the mechanisms of treatment resistance in patients. This could be possible with the advancements in animal models which are further administered with drugs; as the cure for pancreatic cancer partly relies on the elimination of pancreatic CSCs. Since, the genomic make up of each individual is different; individualized or personalized treatment is required to win the battle against cancer. In vitro engineering of mesenchymal stem cells (derived from pancreatic cancer patients) with anti-tumor genes could yield in targeting the cancer cells [88]. Due to the tumor homing capacity of the engineered mesenchymal stem cells, this strategy holds promise towards pancreatic cancer therapy. This strategy could be further expanded to target cancer stem cells which may result in specific treatment options for pancreatic cancer patients.
The authors acknowledge the invaluable support from Ms. Kavita Mallya. We also acknowledge Ms. Stacey L. Therrien for editing this manuscript. The authors on this article were supported by grants from the National Institutes of Health (EDRN UO1CA111294, SPORE P50CA127297, TMEN U54CA163120 and R03 CA167342) and Nebraska Department of Health and Human Services (LB595).
| [1] | Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014, 64:9–29. |
| [2] | Kadera BE, Li L, Toste PA, et al. MicroRNA-21 in pancreatic ductal adenocarcinoma tumor-associated fibroblasts promotes metastasis. PLoS One 2013, 8:e71978. |
| [3] | Hassan MM, Bondy ML, Wolff RA, et al. Risk factors for pancreatic cancer: case-control study. Am J Gastroenterol 2007, 102:2696–707. |
| [4] | Wittel UA, Momi N, Seifert G, Wiech T, Hopt UT, Batra SK. The pathobiological impact of cigarette smoke on pancreatic cancer development (review). Int J Oncol 2012, 41:5–14. |
| [5] | LaFemina J, Roberts PA, Hung YP, et al. Identification of a novel kindred with familial pancreatitis and pancreatic cancer. Pancreatology 2009, 9:273–9. |
| [6] | Permuth-Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Fam Cancer 2009, 8:109–17. |
| [7] | Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev 2014, 28:1–7. |
| [8] | Yonezawa S, Higashi M, Yamada N, Goto M. Precursor lesions of pancreatic cancer. Gut Liver 2008, 2:137–54. |
| [9] | Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008, 3:157–88. |
| [10] | Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am J Surg Pathol 1998, 22:163–9. |
| [11] | Grippo PJ, Tuveson DA. Deploying mouse models of pancreatic cancer for chemoprevention studies. Cancer Prev Res (Phila) 2010, 3:1382–7. |
| [12] | Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L. Mouse models of pancreatic cancer. World J Gastroenterol 2012, 18:1286–94. |
| [13] | Shi G, Zhu L, Sun Y, et al. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology 2009, 136:1368–78. |
| [14] | Bardeesy N, Aguirre AJ, Chu GC, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A 2006, 103:5947–52. |
| [15] | Bardeesy N, Cheng KH, Berger JH, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006, 20:3130–46. |
| [16] | Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 2006, 20:3147–60. |
| [17] | Vaz AP, Ponnusamy MP, Batra SK. Cancer stem cells and therapeutic targets: an emerging field for cancer treatment. Drug Deliv Transl Res 2013, 3:113–20. |
| [18] | Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003, 100:3983–8. |
| [19] | Alvero AB, Chen R, Fu HH, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8:158–66. |
| [20] | Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res 2007, 67:1030–7. |
| [21] | Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol 2008, 26:2862–70. |
| [22] | Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63:5821–8. |
| [23] | Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature 2004, 432:396–401. |
| [24] | Zhang S, Balch C, Chan MW, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68:4311–20. |
| [25] | Todaro M, Francipane MG, Medema JP, Stassi G. Colon cancer stem cells: promise of targeted therapy. Gastroenterology 2010, 138:2151–62. |
| [26] | Van den Broeck A, Vankelecom H, Van DW, et al. Human pancreatic cancer contains a side population expressing cancer stem cell-associated and prognostic genes. PLoS One 2013, 8:e73968. |
| [27] | Bailey JM, Alsina J, Rasheed ZA, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146:245–56. |
| [28] | Li C, Wu JJ, Hynes M, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141:2218–27. |
| [29] | Matsui W, Huff CA, Wang Q, et al. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103:2332–6. |
| [30] | Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1:313–23. |
| [31] | Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol 2009, 11:1487–95. |
| [32] | Herreros-Villanueva M, Zhang JS, Koenig A, et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2013, 2:e61. |
| [33] | Kim SK, Hebrok M. Intercellular signals regulating pancreas development and function. Genes Dev 2001, 15:111–27. |
| [34] | Mimeault M, Batra SK. Concise review: recent advances on the significance of stem cells in tissue regeneration and cancer therapies. Stem Cells 2006, 24:2319–45. |
| [35] | Strobel O, Rosow DE, Rakhlin EY, et al. Pancreatic duct glands are distinct ductal compartments that react to chronic injury and mediate Shh-induced metaplasia. Gastroenterology 2010, 138:1166–77. |
| [36] | Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 2007, 445:886–91. |
| [37] | Samuelson L, Wright N, Gerber DA. Endodermal progenitor cells isolated from mouse pancreas. Stem Cell Discov 2011, 1:44–53. |
| [38] | Zulewski H, Abraham EJ, Gerlach MJ, et al. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001, 50:521–33. |
| [39] | Smukler SR, Arntfield ME, Razavi R, et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell 2011, 8:281–93. |
| [40] | Wang Y, Lanzoni G, Carpino G, et al. Biliary tree stem cells, precursors to pancreatic c ommitted progenitors: evidence for possible life-long pancreatic organogenesis. Stem Cells 2013, 31:1966–79 |
| [41] | Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A 2010, 107:75–80. |
| [42] | Kopp JL, von FG, Mayes E, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22:737–50. |
| [43] | Leach SD. Epithelial differentiation in pancreatic development and neoplasia: new niches for nestin and Notch. J Clin Gastroenterol 2005, 39:S78–S82. |
| [44] | Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11:1282–90. |
| [45] | Hamada S, Masamune A, Takikawa T, et al. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem Biophys Res Commun 2012, 421:349–54. |
| [46] | Storms RW, Trujillo AP, Springer JB, et al. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci U S A 1999, 96:9118–23. |
| [47] | Christ O, Lucke K, Imren S, et al. Improved purification of hematopoietic stem cells based on their elevated aldehyde dehydrogenase activity. Haematologica 2007, 92:1165–72. |
| [48] | Rasheed Z, Wang Q, Matsui W. Isolation of stem cells from human pancreatic cancer xenografts. J Vis Exp 2010, 2169. |
| [49] | Kim MP, Fleming JB, Wang H, et al. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One 2011, 6:e20636. |
| [50] | Jimeno A, Feldmann G, Suarez-Gauthier A, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther 2009, 8:310–4. |
| [51] | Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A 2004, 101:781–6. |
| [52] | Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res 2007, 67:4827–33. |
| [53] | Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2− cancer cells are similarly tumorigenic. Cancer Res 2005, 65:6207–19. |
| [54] | Mimeault M, Batra SK. Characterization of nonmalignant and malignant prostatic stem/progenitor cells by Hoechst side population method. Methods Mol Biol 2009, 568:139–49. |
| [55] | Di Renzo MF, Narsimhan RP, Olivero M, et al. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene 1991, 6:1997–2003. |
| [56] | Gires O, Klein CA, Baeuerle PA. On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer 2009, 9:143. |
| [57] | Vaz A, Ponnusamy MP, Rachagani S, Dey P, Ganti AK, Batra SK. Role of RNA polymerase II associated factor 1 (Paf1/PD2) in facilitating drug resistance of cancer stem cells: a novel cancer stem cell maintenance marker. Br J Cancer 2014, In press:00. |
| [58] | Seguin L, Kato S, Franovic A, et al. An integrin beta-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol 2014, 16:457–468. |
| [59] | Vermeulen L, Todaro M, de Sousa MF, et al. Single-cell cloning of colon cancer stem c ells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci U S A 2008, 105:13427–32. |
| [60] | Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol 2013, 10:607–20. |
| [61] | Curry JM, Thompson KJ, Rao SG, et al. The use of a novel MUC1 antibody to identify cancer stem cells and circulating MUC1 in mice and patients with pancreatic cancer. J Surg Oncol 2013, 107:713–22. |
| [62] | Mimeault M, Johansson SL, Senapati S, Momi N, Chakraborty S, Batra SK. MUC4 down-regulation reverses chemoresistance of pancreatic cancer stem/progenitor cells and their progenies. Cancer Lett 2010, 295:69–84. |
| [63] | Ponnusamy MP, Seshacharyulu P, Vaz A, Dey P, Batra SK. MUC4 stabilizes HER2 expression and maintains the cancer stem cell population in ovarian cancer cells. J Ovarian Res 2011, 4:7. |
| [64] | Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res 2008, 68:2419–26. |
| [65] | Apelqvist A, Li H, Sommer L, et al. Notch signalling controls pancreatic cell differentiation. Nature 1999, 400:877–81. |
| [66] | Bao B, Wang Z, Ali S, et al. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett 2011, 307:26–36. |
| [67] | Abel EV, Kim EJ, Wu J, et al. The notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One 2014, 9:e91983. |
| [68] | Hebrok M. Hedgehog signaling in pancreas development. Mech Dev 2003, 120:45–57. |
| [69] | Rodova M, Fu J, Watkins DN, Srivastava RK, Shankar S. Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS One 2012, 7:e46083. |
| [70] | Murtaugh LC. The what, where, when and how of Wnt/beta-catenin signaling in pancreas development. Organogenesis 2008, 4:81–6. |
| [71] | Zhang Y, Morris JP, Yan W, et al. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res 2013, 73:4909–22. |
| [72] | Stemmer V, de CB, Berx G, Behrens J. Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene 2008, 27:5075–80. |
| [73] | Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep 2013, 3:3230. |
| [74] | Sun L, Mathews LA, Cabarcas SM, et al. Epigenetic regulation of SOX9 by the NF-kappaB signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31:1454–66. |
| [75] | Van den Broeck A, Gremeaux L, Topal B, Vankelecom H. Human pancreatic adenocarcinoma contains a side population resistant to gemcitabine. BMC Cancer 2012, 12:354. |
| [76] | Li Y, VandenBoom TG, Kong D, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res 2009, 69:6704–12. |
| [77] | Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 2009, 69:5820–8. |
| [78] | Mimeault M, Batra SK. Functions of tumorigenic and migrating cancer progenitor cells in cancer progression and metastasis and their therapeutic implications. Cancer Metastasis Rev 2007, 26:203–14. |
| [79] | Jung DE, Wen J, Oh T, Song SY. Differentially expressed microRNAs in pancreatic cancer stem cells. Pancreas 2011, 40:1180–7. |
| [80] | Lonardo E, Cioffi M, Sancho P, et al. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS One 2013, 8:e76518. |
| [81] | Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol 2012, 2012:9506–58. |
| [82] | Rausch V, Liu L, Kallifatidis G, et al. Synergistic activity of sorafenib and sulforaphane abolishes pancreatic cancer stem cell characteristics. Cancer Res 2010, 70:5004–13. |
| [83] | Hage C, Rausch V, Giese N, et al. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis 2013, 4:e627. |
| [84] | Zeng JY, Sharma S, Zhou YQ, et al. Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells. Mol Cancer Ther 2014, 13:37–48. |
| [85] | Li L, Hao X, Qin J, et al. Antibody against CD44s inhibits pancreatic tumor initiation and postradiation recurrence in mice. Gastroenterology 2014, 146:1108–18. |
| [86] | Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res 2011, 31:1105–13. |
| [87] | Balic A, Draeby SM, Trabulo SM, et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther 2014, 13:1–14. |
| [88] | Moniri MR, Dai LJ, Warnock GL. The challenge of pancreatic cancer therapy and novel treatment strategy using engineered mesenchymal stem cells. Cancer Gene Ther 2014, 21:12–23. |
| [89] | Cole MF, Johnstone SE, Newman JJ, Kagey MH, Young RA. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev 2008, 22:746–55. |
| [90] | Nusse R, Fuerer C, Ching W, et al. Wnt signaling and stem cell control. Cold Spring Harb Symp Quant Biol 2008, 73:59–66. |
| [91] | Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med 2009, 9:873–86. |
| [92] | Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425:851–6. |
| [93] | Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455:406–10. |